Loading...
Von Gierke Disease (GSD I)
Von Gierke disease (GSD I) is an autosomal recessive inherited disorder of glycogen metabolism caused by deficiency of the glucose-6-phosphatase (G6Pase) system, resulting in impaired hepatic gluco…
Dr. Lucas Mastropaolo
Von Gierke disease (GSD I) is an autosomal recessive inherited disorder of glycogen metabolism caused by deficiency of the glucose-6-phosphatase (G6Pase) system, resulting in impaired hepatic glucose production via both glycogenolysis and gluconeogenesis. It is subdivided into GSD Ia (~80% of cases; G6PC gene, 17q21) and GSD Ib (~20%; SLC37A4 gene, 11q23). Incidence is approximately 1 in 100,000 births, with higher prevalence in Ashkenazi Jewish populations (~1 in 20,000).[1-2]
1. History
- Key HPI questions: Age of onset, feeding frequency and tolerance, fasting duration before symptoms, history of seizures, growth trajectory, epistaxis or easy bruising, recurrent infections (especially in GSD Ib)
- Symptom characterization: Tremors, irritability, diaphoresis, pallor, or seizures from hypoglycemia; tachypnea from lactic acidosis (may mimic pneumonia); abdominal distension[2]
- Timing/triggers: Symptoms typically appear at 3–4 months of age when feeding intervals increase (e.g., sleeping through the night); intercurrent illness disrupting feeding patterns is a common trigger[2-3]
- Associated symptoms: Failure to thrive, short stature, diarrhea, epistaxis, easy bruising, menorrhagia in adolescent females GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Important negatives: Absence of muscle weakness or elevated CK (distinguishes from GSD III); cognitive development is usually normal unless cerebral damage from recurrent hypoglycemia[2]
2. Alarm Features
- Severe hypoglycemia (BG <40 mg/dL) with seizures, loss of consciousness, or apnea — can occur after only 2–4 hours of fasting[2-3]
- Severe lactic acidosis (lactate often ≥10 mmol/L) with high anion gap metabolic acidosis[2]
- Progressive metabolic acidosis and cardiac dysrhythmia during surgery or prolonged fasting — cardiac arrest has been reported[2]
- Hemorrhage — impaired platelet function and acquired von Willebrand disease; life-threatening menorrhagia, intrahepatic adenoma hemorrhage GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Hepatic adenoma transformation to hepatocellular carcinoma (HCC)[2-3]
- Acute pancreatitis from severe hypertriglyceridemia (>1,000 mg/dL)[2][4-5]
- Sepsis in GSD Ib from neutropenia and neutrophil dysfunction[4]
- Tube dislodgement or pump failure during overnight continuous feeding — can cause fatal hypoglycemia GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
3. Medications
Core treatments
- Uncooked cornstarch (UCCS) — mainstay of therapy; dosed based on hepatic glucose production rate; given between meals and overnight. Glycosade® (extended-release waxy maize cornstarch) allows longer fasting intervals and fewer daily doses GeneReviews® [Internet]. Updated 2021 Oct 14.[3][6]
- Allopurinol or febuxostat for hyperuricemia/gout prevention[2-3]
- ACE inhibitors (captopril, lisinopril) for microalbuminuria and renal protection[2]
- Lipid-lowering agents (statins, fibrates) for persistent hyperlipidemia[2]
- G-CSF for neutropenia in GSD Ib GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Empagliflozin (SGLT2 inhibitor) — emerging first-line therapy for GSD Ib neutropenia; reduces 1,5-anhydroglucitol-6-phosphate accumulation in neutrophils, improves neutrophil function, and allows G-CSF reduction or discontinuation in most patients[7-9]
- Citrate supplementation for hypocitraturia/nephrocalcinosis prevention[2-3]
- DDAVP and antifibrinolytics for bleeding diathesis GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
Contraindicated/avoided medications
- Glucagon — ineffective and worsens lactic acidosis GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Metformin — contraindicated GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Lactated Ringer's — lactate-containing infusions are contraindicated[2-3]
- Combined oral contraceptives (estrogen-containing) — risk of worsening hepatic adenomas GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Amoxicillin/clavulanic acid — increased diarrhea risk; rare idiosyncratic liver failure GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- NSAIDs (ibuprofen) — nephrotoxic, especially with reduced GFR or bleeding diathesis[2]
- Loop diuretics — risk of hypercalciuria[2]
- Beta-blockers — should be avoided for hypertension management GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
4. Diet
- Macronutrient composition: 60–70% complex carbohydrates, 10–15% protein, <30% fat[2]
- Avoid: Sucrose, fructose, galactose, lactose, high-fructose corn syrup, honey, maple syrup, sorbitol — these cannot be converted to free glucose due to G6Pase deficiency and worsen lactic acidosis and hyperlipidemia GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Simple sugars restricted to <5 g/meal and <2 g/snack; certain cheeses and yogurt with low lactose content may be permitted within these limits GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Frequent small meals every 3–4 hours rich in complex carbohydrates (whole grains, legumes, rice) GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Overnight management: Continuous nasogastric/gastrostomy tube feeding in infants (glucose infusion 8–10 mg/kg/min in infants, 6–8 in older children, 3–7 in adults) or uncooked cornstarch at bedtime GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Micronutrient supplementation is essential due to dietary restrictions — calcium, vitamin D, iron, and multivitamins GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Hydration: Adequate fluid intake to prevent nephrolithiasis[2]
5. Review of Systems
- General: Growth retardation, failure to thrive, delayed puberty, fatigue
- GI: Hepatomegaly, abdominal distension, diarrhea, nausea/vomiting (intercurrent illness), abdominal pain (pancreatitis)
- Hematologic: Epistaxis, easy bruising, menorrhagia, recurrent infections (GSD Ib)
- Renal: Hematuria, polyuria (renal tubular dysfunction)
- Musculoskeletal: Bone pain, fractures (osteoporosis), joint pain (gout)
- Skin: Eruptive xanthomas on extensor surfaces and buttocks[2]
- Neurologic: Seizures, developmental delay (if recurrent severe hypoglycemia)
- Oral: Aphthous stomatitis, gingivitis (GSD Ib) GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
6. Collateral History and Family History
- Autosomal recessive inheritance — both parents are carriers; 25% recurrence risk per pregnancy[2]
- Consanguinity increases risk
- Ethnic-specific mutations: Common mutations account for ~90% of disease alleles in certain ethnic groups (Ashkenazi Jewish, Hispanic, Asian populations)[2]
- Siblings should be evaluated with molecular testing or metabolic assessment soon after birth GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Assess family understanding of emergency protocols, dietary compliance, and overnight feeding safety
7. Risk Factors
- Ashkenazi Jewish descent (prevalence ~1/20,000)[2]
- Pan-ethnic but with ethnic-specific common mutations in Caucasians, Hispanics, and Asians[2]
- Poor metabolic control — strongest risk factor for long-term complications including nephropathy, hepatic adenomas, and osteoporosis[2][10]
- Dietary non-adherence — particularly in adolescents and young adults[11]
- Intercurrent illness — disrupts feeding patterns and precipitates metabolic crises[2]
8. Differential Diagnosis
- GSD Type III (Cori disease): Hepatomegaly and hypoglycemia but with ketosis, elevated CK, and muscle involvement; glycemia and lactate are high postprandially and low after fasting (opposite pattern to GSD I)[1-2]
- GSD Types VI and IX: Milder hepatic glycogenoses with ketotic hypoglycemia
- Fatty acid oxidation disorders: Hypoketotic hypoglycemia — included in newborn screening[2]
- Galactosemia: Included in newborn screening; hepatomegaly with reducing substances in urine[2]
- Growth hormone deficiency: Mild hepatomegaly with hypoglycemia — GSD I patients may be misdiagnosed[2]
- Familial hypertriglyceridemia: Extreme hypertriglyceridemia without hepatomegaly or hypoglycemia[11]
- Neuroblastoma with hepatic metastases (Pepper syndrome): Hepatomegaly in infants — ruled out by ultrasound and clinical features[1]
- Niemann-Pick, Gaucher disease: Other hepatomegaly-causing storage disorders
9. Past Medical History
- Previous hypoglycemic episodes, seizures, or hospitalizations for metabolic decompensation
- History of hepatic adenomas (common with increasing age)
- Renal disease — nephrolithiasis, proteinuria, CKD progression
- Gout or gouty arthritis
- Osteoporosis or fractures
- Pancreatitis episodes
- Surgical history — prior liver biopsy, adenoma resection, transplantation
- GSD Ib: history of recurrent infections, inflammatory bowel disease, oral ulcers
- Polycystic ovaries in females[2-3]
10. Physical Exam
- Vital signs: Tachypnea (lactic acidosis), hypotension (if acutely decompensated), hypertension (long-term complication)
- General: Short stature, doll-like facies with full cheeks (cushingoid appearance), thin extremities[2-3]
- Abdomen: Massive hepatomegaly with protuberant abdomen; nephromegaly on palpation; splenomegaly possible in GSD Ib[2-3]
- Skin: Eruptive xanthomas on extensor surfaces and buttocks[2]
- Oral: Aphthous ulcers, gingivitis, periodontitis (GSD Ib) GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Musculoskeletal: No myopathy (distinguishes from GSD III)
- Bleeding signs: Petechiae, ecchymoses, active epistaxis
11. Lab Studies
Diagnostic labs (critical sample during hypoglycemia)
- Blood glucose (<40 mg/dL after 3–4 hour fast), lactate (often ≥10 mmol/L), uric acid (elevated), triglycerides (markedly elevated, serum may appear milky), cholesterol (elevated LDL, low HDL)[2]
- β-hydroxybutyrate (only mildly elevated relative to free fatty acids — hypoketotic)[2]
- Hepatic profile, CK (normal in GSD I), CBC with differential (neutropenia suggests GSD Ib)[2]
Confirmatory testing
- Molecular genetic testing of G6PC (GSD Ia) or SLC37A4 (GSD Ib) — now preferred over liver biopsy[1-2]
- Liver biopsy with enzyme activity measurement rarely needed[1]
Monitoring labs (every 6–12 months)
- Blood glucose, lactate, lipid panel, uric acid, CMP (BUN, creatinine, AST, ALT, albumin, electrolytes), PT/INR, aPTT GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- CBC with differential (every 3 months if on G-CSF) GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Urine: albumin-to-creatinine ratio, calcium, citrate, urinalysis[2]
- Iron studies, ferritin, 25-hydroxyvitamin D, TSH annually GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
12. Imaging
- First-line: Abdominal ultrasound — hepatomegaly, nephromegaly, hepatic adenomas, nephrocalcinosis/nephrolithiasis[2]
Liver surveillance
- Ultrasound every 12–24 months until age 16 GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Liver CT or MRI with contrast every 6–12 months starting at age 16 or earlier if adenomas present — to monitor for malignant transformation GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Renal ultrasound annually beginning at age 10 to assess kidney size and calcifications[2]
- Bone densitometry (DEXA) every 1–2 years for osteoporosis screening GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Brain MRI if concern for cerebral damage from recurrent hypoglycemia — may show occipital horn dilation and subcortical white matter changes GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
13. Special Tests
- Glucagon stimulation test: Absent hyperglycemic response with rise in lactate — historically used but now largely replaced by genetic testing[1]
- Continuous glucose monitoring (CGM): Increasingly used to guide individualized cornstarch dosing, identify nocturnal hypoglycemia, and optimize time-in-range[12]
- Neutrophil function tests: Oxidative burst, chemotaxis (GSD Ib)[7]
- 1,5-anhydroglucitol (1,5-AG) levels: Biomarker for monitoring empagliflozin efficacy in GSD Ib[7][13]
- GFR estimation: Bedside Schwartz (children) or MDRD/CKD-EPI (adults); hyperfiltration defined as eGFR >140 mL/min/1.73 m²[2]
- Bleeding time / platelet function assays preoperatively[2]
14. ECG
- Indications: Preoperative assessment, evaluation for pulmonary hypertension, or if cardiac dysrhythmia suspected during metabolic decompensation
- Pulmonary hypertension is a recognized long-term complication — echocardiography is the primary screening tool[2-3]
- Cardiac dysrhythmia and cardiac arrest have been reported during surgery with progressive metabolic acidosis[2]
- No primary cardiac conduction defect is characteristic of GSD I (unlike GSD III which can cause cardiomyopathy)
15. Assessment
- GSD I is a chronic, multisystem metabolic disorder requiring lifelong dietary management and surveillance
- Severity stratification depends on metabolic control: well-controlled patients have near-normal growth, development, and life span; poorly controlled patients develop progressive hepatic, renal, and skeletal complications[1][3]
- Typical presentation: Infant at 3–4 months with hepatomegaly, hypoglycemia, lactic acidosis, and hyperlipidemia
- Atypical presentations: Rare cases diagnosed in adulthood with adenomas and hyperuricemia; extreme hypertriglyceridemia without classic hypoglycemia[2][11]
- Complications to anticipate: Hepatic adenomas (with HCC risk), progressive CKD, osteoporosis, gout, pancreatitis, pulmonary hypertension, bleeding diathesis, polycystic ovaries[2-3]
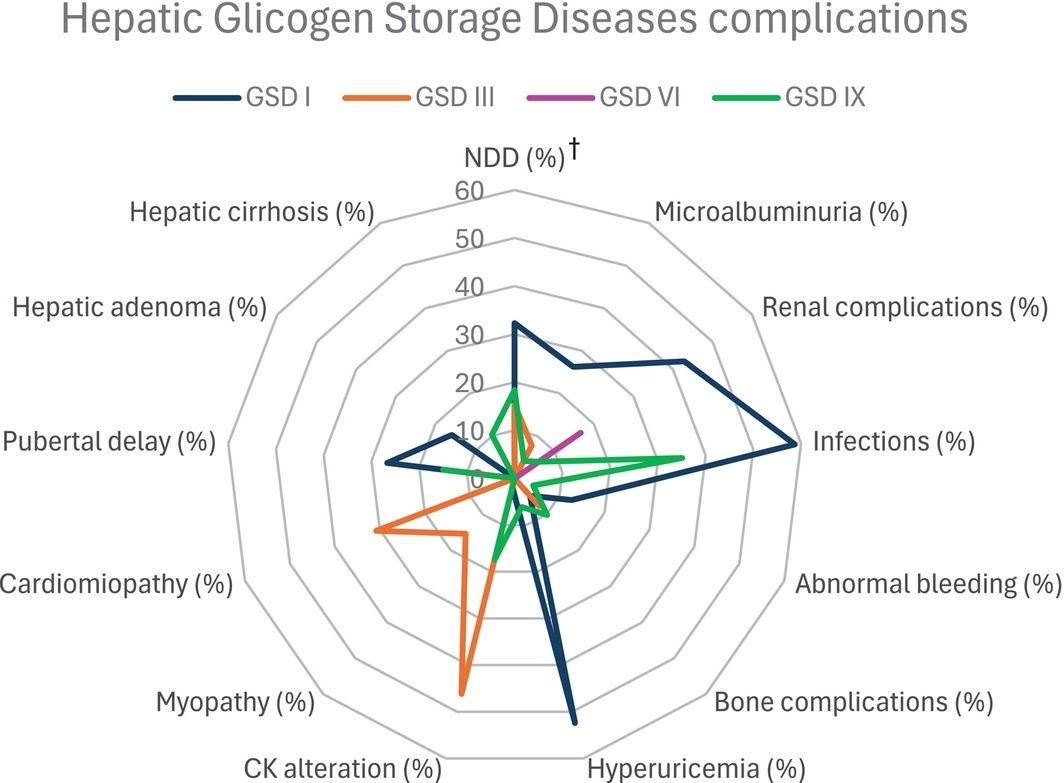
- The following figure illustrates the distinct complication profile of GSD I compared to other hepatic glycogen storage diseases:
16. Treatment Plan
- Initial stabilization (acute hypoglycemia/metabolic crisis):
- IV D10 in 0.5 NS at 1.5–2× maintenance rate; adjust to keep BG ≥70 mg/dL[2]
- Do NOT use glucagon (ineffective, worsens lactic acidosis) GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Do NOT use Lactated Ringer's[2-3]
- Monitor BG, electrolytes, lactate, and acid-base status frequently[2]
Chronic dietary management
- Uncooked cornstarch therapy (Argo® preferred in the US); doses calculated based on hepatic glucose production rate GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Glycosade® for extended overnight fasting[6]
- Frequent meals every 3–4 hours; restrict fructose, galactose, sucrose GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Overnight continuous tube feeding in infants/young children GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Target BG ≥70 mg/dL at all times[2]
Pharmacologic management
- Allopurinol for hyperuricemia[2]
- ACE inhibitors for microalbuminuria/hyperfiltration[2]
- Lipid-lowering agents as needed[2]
- Empagliflozin for GSD Ib neutropenia (starting dose ~0.2 mg/kg; titrate carefully due to hypoglycemia risk)[7][13][15]
- G-CSF for GSD Ib if empagliflozin insufficient GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Vitamin D, calcium, iron supplementation GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
Surgical/interventional
- Hepatic adenoma management: surveillance, percutaneous ethanol injection, radiofrequency ablation, or resection GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Liver transplantation for refractory metabolic control or HCC — corrects hypoglycemia but renal disease may progress[1][3]
- Kidney transplantation for ESRD; combined liver-kidney transplant when indicated[2-3]
Emerging therapy
- DTX401 (AAV8 gene therapy): Phase 1/2 trial showed 68% reduction in daily cornstarch intake and 46% improvement in fasting tolerance at 52 weeks[16]
17. Disposition
Admission criteria
- Inability to maintain oral/NG feedings (vomiting, anorexia from illness)
- Severe hypoglycemia unresponsive to oral glucose
- Lactic acidosis with pH <7.2 or lactate >10 mmol/L
- Acute pancreatitis
- Sepsis or severe infection (especially GSD Ib)
- Preoperative metabolic stabilization (admit day before surgery for IV dextrose)[2]
Discharge criteria
- Tolerating oral feeds with stable BG ≥70 mg/dL
- Lactic acidosis resolved
- Caregivers demonstrate competence with feeding regimen and emergency protocols
Specialist consultation triggers
- Metabolic genetics — all patients at diagnosis and ongoing
- Hepatology — adenoma surveillance, transplant evaluation
- Nephrology — eGFR <60, persistent microalbuminuria/proteinuria, hyperfiltration[2]
- Hematology — severe neutropenia, bleeding diathesis
- GI — inflammatory bowel disease (GSD Ib)
- Endocrinology — growth failure, osteoporosis, polycystic ovaries
18. Follow Up / Return Precautions
- Follow-up timing: Metabolic clinic visits every 3–6 months; more frequent in infancy and during illness GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Home monitoring: Glucometer or CGM for blood glucose; target ≥70 mg/dL[12]
Symptoms requiring immediate reassessment
- Inability to eat or drink for >2–3 hours
- Persistent vomiting or diarrhea
- Seizures or altered mental status
- Rapid breathing (lactic acidosis)
- Fever with neutropenia (GSD Ib)
- Severe abdominal pain (pancreatitis, adenoma hemorrhage)
- Tube dislodgement or pump failure during overnight feeding GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
Patient/caregiver counseling
- Emergency letter should be carried at all times detailing diagnosis, glucose requirements, and contraindicated medications[17]
- Educate on signs of hypoglycemia and metabolic decompensation
- Emphasize that glucagon is ineffective and should not be administered GeneReviews® [Internet]. Updated 2021 Oct 14.[3]
- Dietary adherence is the single most important determinant of long-term outcomes[2][10]
- Expected course: With optimal dietary management and surveillance, patients achieve near-normal growth, puberty, and life span. Long-term hepatic and renal complications remain possible even with good control.[1][3]
References
1. Glucose-6-Phosphatase Deficiency. — Froissart R, Piraud M, Boudjemline AM, et al. Orphanet Journal of Rare Diseases. 2011.
2. Diagnosis and Management of Glycogen Storage Disease Type I: A Practice Guideline of the American College of Medical Genetics and Genomics. — Kishnani PS, Austin SL, Abdenur JE, et al. Genetics in Medicine : Official Journal of the American College of Medical Genetics. 2014.
3. Glycogen Storage Disease Type I. — Bali DS, El-Gharbawy A, Austin S, et al GeneReviews® [Internet]. 2021.
4. Recurrent Pancreatitis and Sepsis in Glycogen Storage Disease Type Ia Caused by Complex Heterozygous Mutations in 2 Sisters: Case Report. — Liu Q, Yu F, Lu H, et al. Medicine. 2022.
5. A Case Report of Acute Pancreatitis With Glycogen Storage Disease Type IA in an Adult Patient and Review of the Literature. — Ai J, He W, Huang X, et al. Medicine. 2020.
6. Short and Long-Term Acceptability and Efficacy of Extended-Release Cornstarch in the Hepatic Glycogen Storage Diseases: Results From the Glyde Study. — Weinstein DA, Jackson RJ, Brennan EA, et al. Orphanet Journal of Rare Diseases. 2024.
7. Treating Neutropenia and Neutrophil Dysfunction in Glycogen Storage Disease Type Ib With an SGLT2 Inhibitor. — Wortmann SB, Van Hove JLK, Derks TGJ, et al. Blood. 2020.
8. Efficacy and Safety of Empagliflozin for Treating Neutropenia and Neutrophil Dysfunction in Paediatric Patients With Glycogen Storage Disease Type Ib: A Systematic Review and Meta-Analysis. — Iwasyk E, Jin R, Tuzzolino F, et al. British Journal of Clinical Pharmacology. 2025.
9. Shifting Towards Empagliflozin First-Line Therapy in Glycogen Storage Disease Type Ib: A Nationwide Real-World Study. — Uçar SK, Mungan NÖ, Gökçay GF, et al. Journal of Inherited Metabolic Disease. 2026.
10. Tight Metabolic Control Plus ACE Inhibitor Therapy Improves GSD I Nephropathy. — Okechuku GO, Shoemaker LR, Dambska M, et al. Journal of Inherited Metabolic Disease. 2017.
11. Infantile Extreme Hypertriglyceridemia Diagnosed as Glycogen Storage Disease Type Ia: A Case Report. — Yuan C, Liu Y, Lyu J, Sun X, Wu J. Medicine. 2026.
12. Continuous Glucose Monitoring-Driven Personalization of Cornstarch Therapy in Glycogen Storage Disease: A Retrospective Analysis. — Ru JH, Ryu JS, Kang Y, Yang S. Yonsei Medical Journal. 2026.
13. Understanding the Role of SGLT2 Inhibitors in Glycogen Storage Disease Type Ib: The Experience of One UK Centre. — Halligan RK, Dalton RN, Turner C, Lewis KA, Mundy HR. Orphanet Journal of Rare Diseases. 2022.
14. Hepatic Glycogen Storage Diseases in Brazil: A Multicenter Study. — Costa MP, Ferreira AR, Rodrigues AT, et al. American Journal of Medical Genetics. Part A. 2026.
15. Efficacy and Safety of Empagliflozin in Glycogen Storage Disease Type Ib: Data From an International Questionnaire. — Grünert SC, Derks TGJ, Adrian K, et al. Genetics in Medicine : Official Journal of the American College of Medical Genetics. 2022.
16. Safety and Efficacy of DTX401, an AAV8-Mediated Liver-Directed Gene Therapy, in Adults With Glycogen Storage Disease Type I a (GSDIa). — Weinstein DA, Derks TG, Rodriguez-Buritica DF, et al. Journal of Inherited Metabolic Disease. 2025.
17. Glycogen Storage Diseases. — Hannah WB, Derks TGJ, Drumm ML, et al. Nature Reviews. Disease Primers. 2023.