Autoimmune Hemolytic Anemia
Autoimmune hemolytic anemia is an acquired condition in which autoantibodies target red blood cell surface antigens, leading to premature RBC destruction. It is classified by antibody thermal react…
Autoimmune hemolytic anemia is an acquired condition in which autoantibodies target red blood cell surface antigens, leading to premature RBC destruction. It is classified by antibody thermal reactivity: warm AIHA (60–70% of cases, IgG-mediated, extravascular hemolysis) and cold agglutinin disease (20–25%, IgM-mediated, complement-driven intravascular hemolysis), with rarer mixed and paroxysmal cold hemoglobinuria subtypes. [1-2] Incidence is approximately 0.8–3 per 100,000/year, and ~50% of warm AIHA cases are secondary to an underlying condition. [3-4]
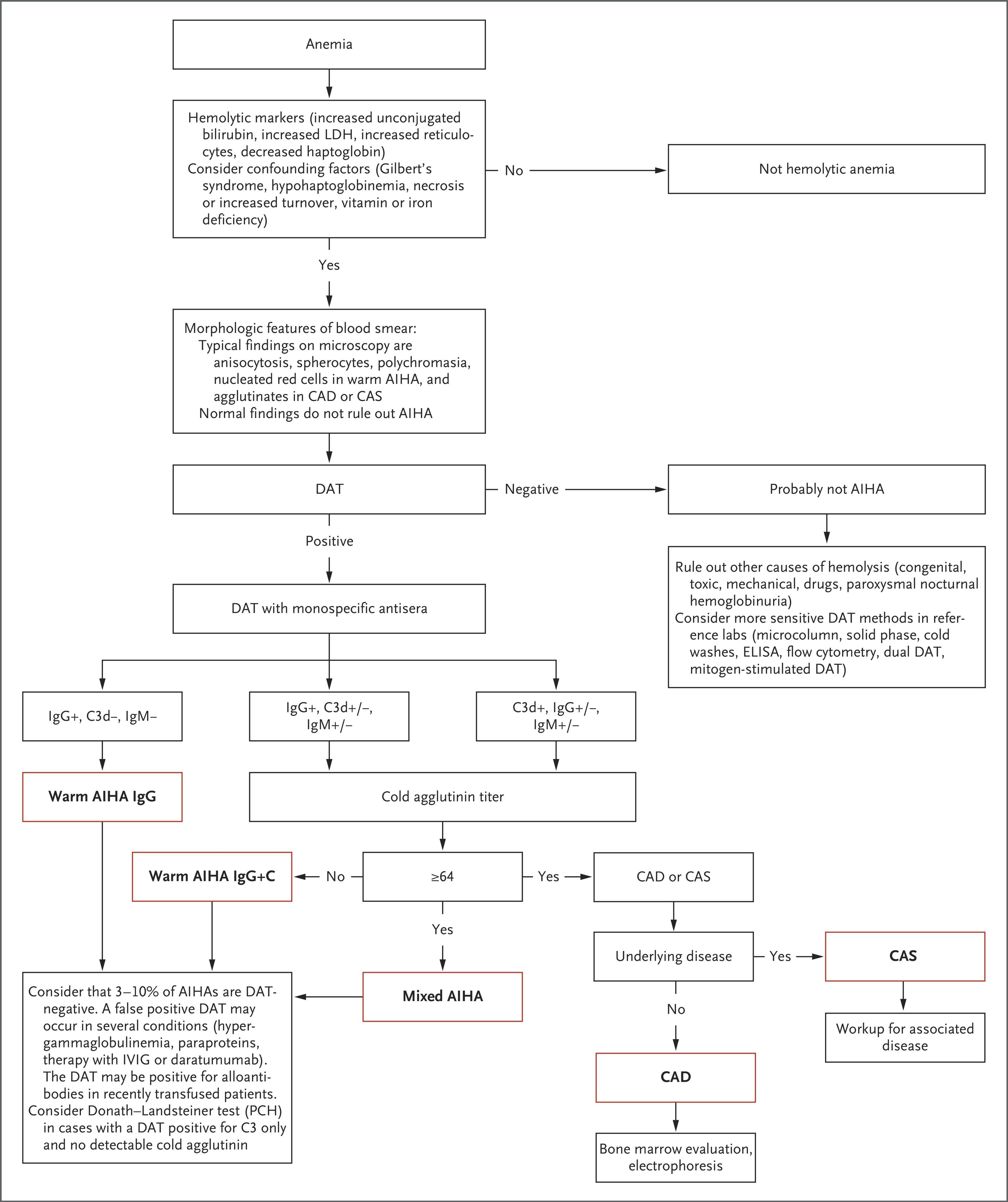
The following diagnostic algorithm from the NEJM provides a systematic approach to AIHA subtype classification:
1. History
- Onset and tempo of symptoms: acute vs. insidious fatigue, exertional dyspnea, palpitations [3]
- Dark or "soy sauce-colored" urine (hemoglobinuria) — suggests intravascular hemolysis [6]
- Jaundice — timing and progression
- Cold-triggered symptoms: acrocyanosis, Raynaud-like phenomena, worsening with cold exposure (suggests cold AIHA) [3]
- Recent infections (viral illness, Mycoplasma, EBV — triggers for cold agglutinin syndrome; SARS-CoV-2) [4-5]
- Medication history: recent antibiotics (cephalosporins, piperacillin), checkpoint inhibitors, fludarabine, NSAIDs [5]
- Prior blood transfusions and any transfusion reactions
- History of autoimmune disease (SLE, RA, scleroderma) or lymphoproliferative disorder (CLL, NHL) [3]
- Prior episodes of anemia or hemolysis; chronicity and relapsing pattern [2]
2. Alarm Features
- Hemoglobin <6 g/dL — requires emergent transfusion [3]
- Brisk intravascular hemolysis with chest pain, lethargy, confusion — medical emergency [3]
- Tachycardia, hypotension, or signs of cardiovascular decompensation
- Rapidly dropping hemoglobin unresponsive to transfusion [7]
- Dark urine (hemoglobinuria) with hyperkalemia — risk of renal injury
- Concurrent thrombocytopenia (Evans syndrome) — raises concern for TTP/HUS overlap or underlying immunodeficiency [8]
- Venous thromboembolism: absolute risk of VTE is 15–30% in warm AIHA [3]
3. Medications
- Causative agents: ceftriaxone, other cephalosporins, piperacillin, NSAIDs, penicillins, fludarabine, checkpoint inhibitors (PD-1 inhibitors), methyldopa [5]
- First-line treatment: Prednisone 1 mg/kg/day (60–100 mg/day); ~80% initial response rate but only 30–40% sustained remission at 1 year [5]
- Second-line: Rituximab 375 mg/m² weekly × 4 cycles or 1000 mg × 2 doses (2-week interval); 70–80% response rate [3][5]
- IVIG: Consider in severe cases not responding within 7 days, or as bridge therapy [8]
- Prophylactic anticoagulation: Recommended during severe hemolysis given high VTE risk [2-3]
- Folic acid supplementation: Standard supportive measure given increased RBC turnover [4]
- Recombinant erythropoietin: When reticulocytopenia or inadequate marrow compensation is present [2]
- Cold AIHA: Steroids are largely ineffective chronically; rituximab is first-line; sutimlimab (anti-C1s complement inhibitor) is FDA-approved for hemolysis in cold agglutinin disease [9-10]
- Contraindicated/caution: Avoid fludarabine monotherapy in CLL patients (exacerbates AIHA); avoid cold exposure in cold AIHA [3][5]
4. Diet
- No specific dietary triggers for AIHA
- Folic acid-rich foods (leafy greens, legumes) to support erythropoiesis
- Adequate hydration, especially during brisk hemolysis, to maintain renal perfusion and urine output
- Iron supplementation is generally not indicated unless concurrent iron deficiency is documented (hemolysis itself does not cause iron loss unless significant hemoglobinuria)
5. Review of Systems
- Constitutional: Fatigue, malaise, fever (may suggest underlying infection or lymphoma)
- HEENT: Scleral icterus, pallor of conjunctivae/mucous membranes
- Cardiovascular: Dyspnea on exertion, palpitations, chest pain, peripheral edema [3]
- GI: Abdominal pain (splenic infarction/congestion), dark urine, change in stool color
- Musculoskeletal: Joint pain (SLE, RA as secondary causes)
- Skin: Rash (lupus), acrocyanosis or livedo reticularis (cold AIHA), petechiae (Evans syndrome)
- Neurologic: Confusion, lethargy (severe anemia or brisk hemolysis) [3]
- Lymphatic: Lymphadenopathy, night sweats, weight loss (lymphoproliferative disease)
6. Collateral History and Family History
- Family history of autoimmune diseases (SLE, thyroid disease, other cytopenias)
- Family history of hereditary hemolytic anemias (spherocytosis, G6PD deficiency) to exclude mimics
- History of autoimmune lymphoproliferative syndrome (ALPS) — germline FAS, FASL, CASP10 mutations predispose to AIHA [3]
- Common variable immunodeficiency (CVID) — associated with AIHA [3]
- Social history: occupational cold exposure (relevant for cold AIHA), HIV risk factors
7. Risk Factors
- Autoimmune diseases: SLE, rheumatoid arthritis, scleroderma [3]
- Lymphoproliferative disorders: CLL, non-Hodgkin lymphoma, Hodgkin lymphoma [3][5]
- Immunodeficiency: CVID, ALPS, post-transplant (HSCT or solid organ) [3]
- Infections: HIV, EBV, Mycoplasma pneumoniae, SARS-CoV-2, babesiosis (especially post-splenectomy) [3][5]
- Medications: Checkpoint inhibitors, cephalosporins, fludarabine, piperacillin [5]
- Age: More common after age 50; bimodal distribution in some series [4]
- Monoclonal gammopathy: Present in >20% of warm AIHA patients, more than 5× the expected rate [3]
8. Differential Diagnosis
- Thrombotic thrombocytopenic purpura (TTP) / Hemolytic uremic syndrome (HUS): Schistocytes on smear, thrombocytopenia, renal failure; DAT-negative
- Paroxysmal nocturnal hemoglobinuria (PNH): Flow cytometry for GPI-anchored proteins; DAT-negative
- Hereditary spherocytosis: Family history, osmotic fragility test; DAT-negative
- G6PD deficiency: Oxidative stress triggers, bite cells/Heinz bodies; DAT-negative
- Drug-induced hemolytic anemia: Temporal relationship with offending drug; may be DAT-positive [5]
- Delayed hemolytic transfusion reaction: Recent transfusion history; DAT-positive with alloantibody
- Microangiopathic hemolytic anemia (MAHA/DIC): Schistocytes, abnormal coagulation panel; DAT-negative [11]
- Sickle cell disease / Hemoglobinopathies: Hemoglobin electrophoresis
- Evans syndrome: AIHA + immune thrombocytopenia ± autoimmune neutropenia [8]
Key distinguishing feature: A positive DAT strongly supports AIHA over non-immune causes, though DAT-negative AIHA occurs in 3–10% of cases. [5]
9. Past Medical History
- Prior episodes of hemolytic anemia or documented AIHA (relapsing course is common)
- Known autoimmune conditions (SLE, thyroiditis, inflammatory bowel disease)
- Lymphoproliferative disorders or prior chemotherapy
- Prior splenectomy (affects management options and increases babesiosis risk)
- History of VTE (AIHA independently increases thrombotic risk) [3]
- Transfusion history and known alloantibodies
- Immunodeficiency syndromes
10. Physical Exam
- Vital signs: Tachycardia, tachypnea, hypotension (in severe/acute hemolysis)
- General: Pallor, jaundice/icterus proportional to severity [3]
- HEENT: Scleral icterus, conjunctival pallor
- Cardiovascular: Flow murmur, elevated JVP, peripheral edema (high-output failure) [3]
- Abdomen: Splenomegaly (especially with underlying lymphoproliferative disease); hepatomegaly [3]
- Skin: Acrocyanosis, livedo reticularis (cold AIHA); petechiae/purpura (Evans syndrome); rash (SLE)
- Lymph nodes: Lymphadenopathy (suggests lymphoma, CLL, or infection)
- Extremities: Peripheral cyanosis with cold exposure (cold agglutinin disease)
11. Lab Studies
Pearl: A negative DAT does not rule out AIHA — occurs in 3–10% of cases. Consider monospecific DAT (including anti-IgA) and more sensitive methods (flow cytometry DAT) if clinical suspicion is high. [5][10]
12. Imaging
- CT chest/abdomen/pelvis: Indicated to evaluate for lymphadenopathy, splenomegaly, or underlying lymphoproliferative/solid malignancy as a secondary cause [5]
- Ultrasound abdomen: Can assess splenomegaly and hepatomegaly
- Imaging is not required for diagnosis of AIHA itself but is important for identifying secondary causes
- Chest imaging if VTE is suspected (CT pulmonary angiography) given the 15–30% VTE risk [3]
13. Special Tests
- Donath-Landsteiner test: Detects biphasic IgG autoantibody in paroxysmal cold hemoglobinuria (mainly in children) [5]
- Flow cytometry (peripheral blood): Screen for CLL, lymphoma, or PNH [5]
- Bone marrow biopsy: Indicated in cold agglutinin disease (to identify clonal B-cell disorder); in warm AIHA if treatment fails or bone marrow disorder is suspected [5]
- Immunoglobulin quantification: Detect CVID (hypogammaglobulinemia) or monoclonal gammopathy [5]
- Viral serologies: HIV, hepatitis B/C, EBV, CMV as clinically indicated
- ANA, dsDNA, complement levels: If SLE or other connective tissue disease is suspected
- ADAMTS13 activity: If TTP is in the differential (thrombocytopenia + MAHA)
14. ECG
- Indications: Severe anemia (Hb <7 g/dL), chest pain, tachycardia, elderly patients, known cardiac disease
- Expected findings: Sinus tachycardia, ST-T wave changes from demand ischemia
- Dangerous patterns: ST elevation or depression suggesting ACS in the setting of severe anemia; new arrhythmias
- Continuous telemetry monitoring warranted in hemodynamically significant anemia
15. Assessment
Severity stratification: [2][5]
- Mild/compensated: Hemoglobin near normal, reticulocytosis compensates for hemolysis, minimal symptoms
- Moderate: Hb 8–10 g/dL, symptomatic fatigue/dyspnea, requires treatment
- Severe: Hb <8 g/dL — international consensus recommends adding rituximab to steroids upfront [5]
- Life-threatening: Hb <6 g/dL, hemodynamic instability, brisk intravascular hemolysis with chest pain/confusion — medical emergency [3]
Warm AIHA carries a mortality rate of up to 22% and has a chronic, relapsing course. [7] More than 50% of patients require blood transfusion at presentation. [3] Always evaluate for secondary causes, as ~50% of warm AIHA is associated with an underlying condition. [3][5]
16. Treatment Plan
Initial Stabilization (ED/Acute)
- Transfuse ABO- and RhD-matched blood if Hb <6 g/dL; goal Hb >7 g/dL. Do not withhold transfusion due to crossmatch difficulties — "least incompatible" blood is acceptable in emergencies [3]
- Monitor for transfusion reactions (fever, chills, worsening hemoglobinuria) [3]
- Check potassium (hemolysis-related hyperkalemia) [7]
- Prednisone 1 mg/kg/day (60–100 mg/day) for 2–3 weeks, then taper over 3–6 months
- Add rituximab upfront if Hb <8 g/dL (severe disease) — two RCTs showed doubling of long-term responses with combination therapy [5]
- IVIG if no response within 7 days and severe anemia [8]
- Prophylactic anticoagulation during active severe hemolysis [2]
- Folic acid 1 mg daily
Warm AIHA — Second-Line [3][5]
- Rituximab (if not given first-line): 375 mg/m² weekly × 4 or 1000 mg × 2 doses
- Response in 70–80%, but ~30% relapse within 3 years
Warm AIHA — Third-Line and Beyond [3][5]
- Splenectomy (~70% response, but infection/thrombosis risks; increasingly deferred)
- Immunosuppressants: mycophenolate mofetil, azathioprine, cyclosporine, cyclophosphamide
- Emerging agents: fostamatinib, rilzabrutinib, nipocalimab, sovleplenib [2][13]
Cold Agglutinin Disease [10]
- Avoid cold exposure
- Steroids are largely ineffective long-term
- Rituximab is first-line for treatment-requiring CAD
- Sutimlimab (Enjaymo, anti-C1s) — FDA-approved for hemolysis in CAD [9]
- Rituximab + bendamustine for refractory cases
Emergency/Refractory [5]
- High-dose IV methylprednisolone, IVIG, plasma exchange
- Complement C1 inhibition
- Bortezomib + dexamethasone in refractory cases
17. Disposition
- Admit: Hb <7 g/dL, hemodynamic instability, brisk intravascular hemolysis, need for transfusion, new diagnosis requiring workup, severe symptoms (chest pain, confusion, dyspnea at rest) [3][7]
- Observation: Moderate anemia (Hb 7–10 g/dL) with active hemolysis, pending response to initial steroids
- Discharge: Stable hemoglobin, mild/compensated hemolysis, established diagnosis with outpatient hematology follow-up arranged, reliable patient
- Hematology consultation: All new diagnoses of AIHA; urgent for severe or refractory cases [12]
- Consider ICU: Life-threatening anemia (Hb <5 g/dL), hemodynamic compromise, multiorgan dysfunction
18. Follow Up / Return Precautions
- Follow-up timing: Hematology within 1–2 weeks of discharge; CBC and hemolysis labs (LDH, haptoglobin, reticulocyte count, bilirubin) at each visit [3]
- Steroid taper monitoring: Reassess response at 2–3 weeks; taper over 3–6 months; monitor for steroid side effects (glucose, blood pressure, bone density) [5]
- Monitor for VTE: Counsel on symptoms of DVT/PE; consider prophylactic anticoagulation during active hemolysis [3]
- Return precautions: Worsening fatigue, dyspnea at rest, chest pain, dark urine, fever, new bleeding or bruising (Evans syndrome), syncope
- Long-term counseling: AIHA is often a chronic, relapsing condition; additional therapies may be needed. Workup for secondary causes should be completed once stable [3][5]
- Expected course: Initial steroid response in ~80%, but sustained remission in only 30–40% at 1 year; relapse is common and should not be unexpected [5]
- Vaccinations: If splenectomy is planned, ensure pneumococcal, meningococcal, and Haemophilus influenzae type b vaccines are administered ≥2 weeks prior [5]
References
1. Autoimmune Haemolytic Anaemias. — Michel M, Crickx E, Fattizzo B, Barcellini W. Nature Reviews. Disease Primers. 2024.
2. Management of Autoimmune Hemolytic Anemia. — Barcellini W, Fattizzo B. Hematology. American Society of Hematology. Education Program. 2025.
3. Warm Autoimmune Hemolytic Anemia. — Brodsky RA. The New England Journal of Medicine. 2019.
4. Disease-Modifying Treatments for Primary Autoimmune Haemolytic Anaemia. — Liu AP, Cheuk DK. The Cochrane Database of Systematic Reviews. 2021.
5. Autoimmune Hemolytic Anemias. — Berentsen S, Barcellini W. The New England Journal of Medicine. 2021.
6. Case Report: Severe Autoimmune Hemolytic Anemia in an Elderly Patient Caused by Warm-Reactive IgG and IgA Autoantibodies. — Xia W, Lu J, Hou J, et al. Frontiers in Immunology. 2025.
7. Hematologic Emergencies: Recognition and Initial Management. — Jones DE, Walker JJ, Abellada AMP. American Family Physician. 2024.
8. Diagnosis and Management of Evans Syndrome in Adults: First Consensus Recommendations. — Fattizzo B, Marchetti M, Michel M, et al. The Lancet. Haematology. 2024.
9. FDA Orange Book. — FDA Orange Book. 2026.
10. Autoimmune Hemolytic Anemia: New Frontiers in Diagnosis and Therapy. — Barcellini W, Fattizzo B. Blood Reviews. 2026.
11. Hemolytic Anemia: Evaluation and Differential Diagnosis. — Phillips J, Henderson AC. American Family Physician. 2018.
12. Management of Immune Checkpoint Inhibitor-Related Toxicities. — Updated 2025-10-23. National Comprehensive Cancer Network.
13. Sovleplenib in Patients With Primary or Secondary Warm Autoimmune Haemolytic Anaemia: Results From Phase 2 of a Randomised, Double-Blind, Placebo-Controlled, Phase 2/3 Study. — Zhao X, Sun J, Zhang Z, et al. The Lancet. Haematology. 2025.