Immune Thrombocytopenic Purpura (ITP)
ITP is the most common cause of isolated thrombocytopenia, an acquired autoimmune disorder in which autoantibodies target platelets for premature destruction, primarily in the spleen and liver. [1-…
ITP is the most common cause of isolated thrombocytopenia, an acquired autoimmune disorder in which autoantibodies target platelets for premature destruction, primarily in the spleen and liver. [1-2] It is a diagnosis of exclusion — defined as a platelet count <100 × 10³/μL with no other identifiable cause of thrombocytopenia. [1] Incidence is 2–5 per 100,000 person-years, with peaks at ages 20–30 (slight female predominance) and >60 years (equal sex distribution). [1][3]
1. History
- Onset and timing: Acute vs. gradual onset; in children, often sudden and self-limited; in adults, typically insidious and chronic (up to 70% develop chronic ITP) [1][4]
- Bleeding symptoms: Easy bruising, petechiae, purpura, epistaxis, gum bleeding, menorrhagia, hematuria, GI bleeding [4-5]
- Severity and progression: Quantify bleeding frequency, volume, and impact on daily activities
- Preceding illness: Recent viral URI (especially in children), vaccination history (MMR, varicella, COVID-19, influenza) [4][6]
- Medication review: New medications within 3–10 days (quinine/quinidine, acetaminophen, carbamazepine, rifampicin, vancomycin, abciximab, TMP-SMX); OTC drugs, herbals, supplements [1][7]
- Anticoagulant/antiplatelet use: Increases bleeding risk at any platelet level [8]
- Important negatives: Absence of fever, weight loss, night sweats, bone pain, joint symptoms, lymphadenopathy, hepatosplenomegaly — these suggest alternative diagnoses [1]
2. Alarm Features
- Platelet count <10 × 10³/μL — high risk of spontaneous, life-threatening bleeding [2]
- Intracranial hemorrhage (ICH) — rare but the most feared complication; presents with severe headache, altered mental status, focal neurologic deficits [4]
- Active serious bleeding: GI hemorrhage, hemodynamic instability, hematuria with clots
- Wet purpura (oral blood blisters) — marker of more severe mucosal bleeding risk [8]
- Neurologic symptoms — must rule out TTP (schistocytes, elevated LDH, low ADAMTS13) [1][9]
- Fever + thrombocytopenia — consider TTP/HUS, DIC, sepsis [6]
- Concurrent anemia with schistocytes — suggests thrombotic microangiopathy, not ITP [6][9]
3. Medications
First-line treatments
- Corticosteroids: Prednisone 1–2 mg/kg/day for 1–2 weeks then taper, or dexamethasone 40 mg/day × 4 days (up to 4 cycles); ~80% initial response rate, but >50% relapse on taper [1][3]
- IVIG: 1 g/kg × 1–2 days; raises platelets within 1–4 days in ~80% of patients; effect transient (1–2 weeks); indicated for active serious bleeding or platelets <10 × 10³/μL [1]
- Tranexamic acid: Adjunct for mucosal bleeding [10]
Second-line treatments
- TPO-receptor agonists: Romiplostim (1–10 μg/kg SC weekly), eltrombopag (25–75 mg PO daily), avatrombopag (5–40 mg PO daily) — for ITP refractory to corticosteroids [1][11]
- Rituximab: 375 mg/m² IV weekly × 4 weeks; sustained response in ~60% at 6 months, ~30% at 2 years [1]
- Fostamatinib: 100–150 mg PO BID for chronic ITP refractory to prior treatment [1][11]
- Splenectomy: Reserved for chronic ITP (>12 months) refractory to medical therapy [1]
Medications to AVOID in ITP
- NSAIDs and aspirin — impair platelet function and increase bleeding risk
- Anticoagulants — discontinue during active bleeding; reintroduce only when platelets >50 × 10³/μL [1]
- Drugs known to cause thrombocytopenia: Quinine, quinidine, heparin, carbamazepine, vancomycin, rifampicin, TMP-SMX [1][7]
Cautions with treatment
- Prolonged corticosteroid use should be avoided (diabetes, osteoporosis, infection risk) [1]
- Eltrombopag: take 4 hours after and 2 hours before calcium/iron-containing foods; monitor LFTs [1]
- Rituximab: screen for HBV before use; risk of PML (very rare) [1]
4. Diet
- No specific dietary triggers for ITP
- Avoid alcohol — can independently suppress platelet production and worsen thrombocytopenia (3–43% of alcohol-dependent individuals develop thrombocytopenia) [6]
- Eltrombopag interaction: Must be taken on an empty stomach; avoid dairy, calcium, iron, and antacids within the specified window [1]
- Adequate hydration and nutrition to support marrow function
5. Review of Systems
- Skin: Petechiae, purpura, ecchymoses, new or worsening bruising
- HEENT: Epistaxis, gum bleeding, oral blood blisters (wet purpura), visual changes
- GI: Hematemesis, melena, hematochezia, abdominal pain
- GU: Hematuria, menorrhagia
- Neuro: Headache, confusion, focal deficits (red flag for ICH or TTP)
- Constitutional: Fever, weight loss, night sweats (suggest malignancy, infection, or TTP — atypical for primary ITP) [1][6]
- MSK: Arthralgias, joint swelling (suggest SLE or other autoimmune disease) [1]
- Infectious: Recent illness, travel history, high-risk behaviors (HIV, hepatitis) [1][6]
6. Collateral History and Family History
- Family history of bleeding disorders or thrombocytopenia — consider congenital thrombocytopenias (Bernard-Soulier, MYH9-related disorders) [1]
- Family history of autoimmune disease — SLE, rheumatoid arthritis, thyroid disease increases likelihood of secondary ITP [1]
- Social context: Ability to recognize bleeding symptoms, access to emergency care, medication compliance, alcohol use [6][12]
- Collateral from prior labs: Obtain previous CBC results to distinguish acute vs. chronic thrombocytopenia [6]
7. Risk Factors
- Age: Bimodal — young adults (20–30) and older adults (>60) [1]
- Sex: Slight female predominance in younger patients; equal in older adults [1]
- Autoimmune comorbidities: SLE, rheumatoid arthritis, antiphospholipid syndrome, thyroid disease [1]
- Infections: HIV, HCV, HBV, CMV, EBV, H. pylori — all associated with secondary ITP [1]
- Lymphoproliferative disorders: CLL, Hodgkin lymphoma [1]
- Immunodeficiency syndromes: CVID, autoimmune lymphoproliferative syndrome [1]
- Recent vaccination (rare trigger) [6]
- Pregnancy — must distinguish from gestational thrombocytopenia and preeclampsia/HELLP [13]
8. Differential Diagnosis
Dangerous cannot-miss diagnoses
- Thrombotic thrombocytopenic purpura (TTP): Neurologic symptoms, MAHA (schistocytes), elevated LDH, low ADAMTS13 (<10%) [1][9]
- Hemolytic uremic syndrome (HUS): Renal failure predominant, MAHA [1]
- Disseminated intravascular coagulation (DIC): Elevated PT/PTT, low fibrinogen, elevated D-dimer [6]
- Heparin-induced thrombocytopenia (HIT): Thrombosis + heparin exposure [1]
- Acute leukemia/MDS: Other cytopenias, blasts on smear [1]
Common mimics
- Pseudothrombocytopenia: EDTA-induced platelet clumping; repeat in citrate tube [1]
- Drug-induced thrombocytopenia: Resolves 7–10 days after drug withdrawal [6]
- Gestational thrombocytopenia: Mild (usually >70 × 10³/μL), third trimester, resolves postpartum [13]
- Liver disease/hypersplenism: Hepatosplenomegaly, abnormal LFTs [1]
- Evans syndrome: ITP + DAT-positive hemolytic anemia [1]
9. Past Medical History
- Prior episodes of thrombocytopenia or ITP (relapse vs. chronic)
- Autoimmune diseases: SLE, RA, antiphospholipid syndrome, thyroid disease [1]
- Chronic infections: HIV, hepatitis B/C [1]
- Lymphoproliferative disorders [1]
- Splenectomy history — affects treatment options
- Prior response to corticosteroids or IVIG — guides current management
- Surgical history: Bariatric surgery (nutritional deficiencies), valve replacement (mechanical destruction) [6]
10. Physical Exam
Vital signs: Tachycardia and hypotension suggest active hemorrhage
Focused exam
- Skin: Petechiae (dependent areas, pressure points), purpura, ecchymoses — quantify size and distribution [6]
- Oral cavity: Wet purpura (hemorrhagic bullae on buccal mucosa), gum bleeding — indicates higher bleeding risk [5][8]
- Fundoscopic exam: Retinal hemorrhages (concern for ICH risk)
- Lymph nodes: Lymphadenopathy suggests lymphoproliferative disorder, infection, or SLE — atypical for primary ITP [1][6]
- Abdomen: Hepatosplenomegaly — suggests liver disease, lymphoma, or portal hypertension; spleen is NOT typically enlarged in primary ITP [1][6]
- Neurologic: Mental status, focal deficits — if present, urgently evaluate for ICH or TTP [6]
Expected findings in ITP: Isolated mucocutaneous bleeding signs with an otherwise normal exam. Any systemic findings (lymphadenopathy, splenomegaly, fever) should prompt evaluation for secondary causes. [1]
11. Lab Studies
Initial workup
- CBC with differential: Isolated thrombocytopenia; other cell lines normal [1]
- Peripheral blood smear: Reduced platelets, may show large platelets (not pathognomonic); no schistocytes, blasts, or dysplastic changes [1]
- Reticulocyte count: Normal (unless concurrent bleeding)
- Coagulation studies (PT/INR, PTT, fibrinogen): Normal in ITP; abnormal suggests DIC [6]
To rule out secondary causes
- Hemolysis panel: LDH, haptoglobin, indirect bilirubin, DAT (Coombs test) — to exclude TTP, Evans syndrome [1][6]
- Viral serologies: HIV, HBV, HCV [1][6]
- H. pylori testing: Stool antigen or breath test [1][6]
- Renal and hepatic function tests [6]
- ANA, antiphospholipid antibodies — if autoimmune disease suspected [1]
- Immunoglobulin levels — if immunodeficiency suspected [1]
- Thyroid function tests — when clinically indicated [6]
Not routinely recommended
- Antiplatelet antibody testing: Detected in only 50–60% of ITP patients; not recommended in diagnostic workup [1]
- Bone marrow biopsy: Not diagnostic; reserved for atypical presentations, other cytopenias, or treatment-refractory cases [1]
12. Imaging
- Not routinely required for diagnosis of primary ITP
- Abdominal ultrasound: Assess for hepatosplenomegaly if suspected on exam; splenomegaly suggests alternative diagnosis (portal hypertension, lymphoma) [1]
- CT chest/abdomen/pelvis: Only if lymphoproliferative disorder or solid malignancy suspected (weight loss, night sweats, lymphadenopathy) [1]
- CT head (non-contrast): Urgently indicated if any neurologic symptoms to rule out intracranial hemorrhage [4]
13. Special Tests
- Immature platelet fraction (IPF%) or reticulated platelets: Elevated in ITP (increased platelet turnover); helps differentiate from hypoproductive causes [14]
- Plasma thrombopoietin (TPO) levels: Normal or slightly increased in ITP; markedly elevated in aplastic anemia [14]
- ADAMTS13 activity: Order if TTP is in the differential (thrombocytopenia + MAHA); <10% confirms TTP [9]
- Flow cytometry: If concern for leukemia or lymphoproliferative disorder [1]
- Bone marrow aspirate/biopsy: Only for atypical features, other cytopenias, age >60 (to exclude MDS), or treatment failure [1]
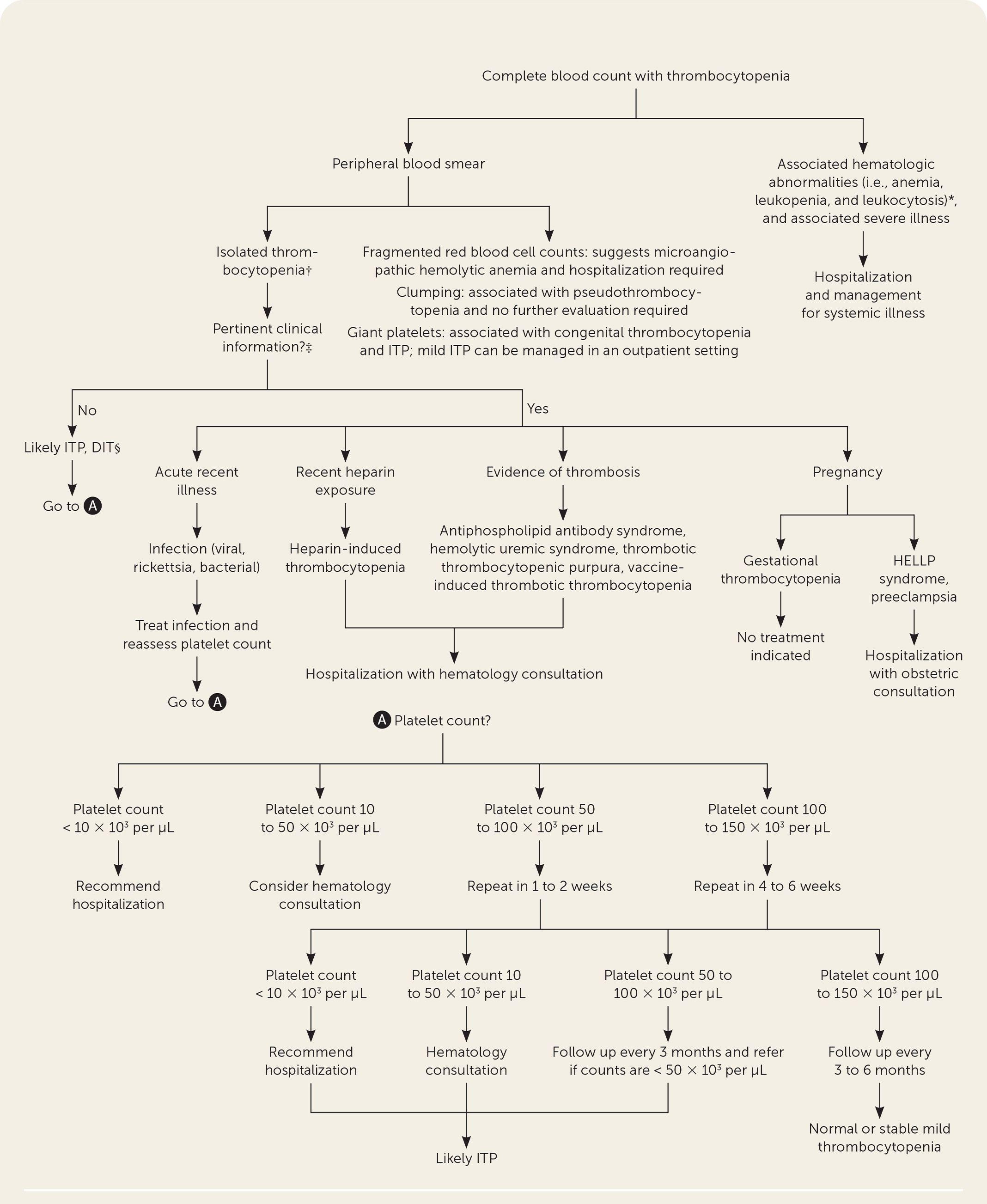
The following figure illustrates the evaluation algorithm for thrombocytopenia:
14. ECG
- Not routinely indicated for ITP itself
- Consider ECG if:
- Hemodynamically significant bleeding (tachycardia, hypotension)
- Concurrent chest pain or dyspnea (pericardial effusion from autoimmune disease)
- Evaluating for TTP (cardiac ischemia can occur) [9]
- No ITP-specific ECG patterns
15. Assessment
Classification by duration: [15]
- Newly diagnosed: <3 months from diagnosis
- Persistent: 3–12 months
- Chronic: >12 months
Severity stratification
- Platelets ≥30 × 10³/μL, no bleeding → observation may be appropriate [8]
- Platelets <30 × 10³/μL → corticosteroids recommended regardless of bleeding [3][8]
- Platelets <20 × 10³/μL → consider hospitalization for newly diagnosed patients [2][12]
- Platelets <10 × 10³/μL → high risk of spontaneous bleeding; add IVIG [1-2]
Key clinical pearls
- Only ~5% of ITP patients present with severe bleeding, but ~15% develop bleeding requiring hospitalization within 5 years [1]
- VTE risk is twice that of the general population despite low platelets [1]
- Fatigue and impaired quality of life are common even without bleeding [1]
- Chronic ITP develops in up to 70% of adults; spontaneous remission can occur years later [1]
16. Treatment Plan
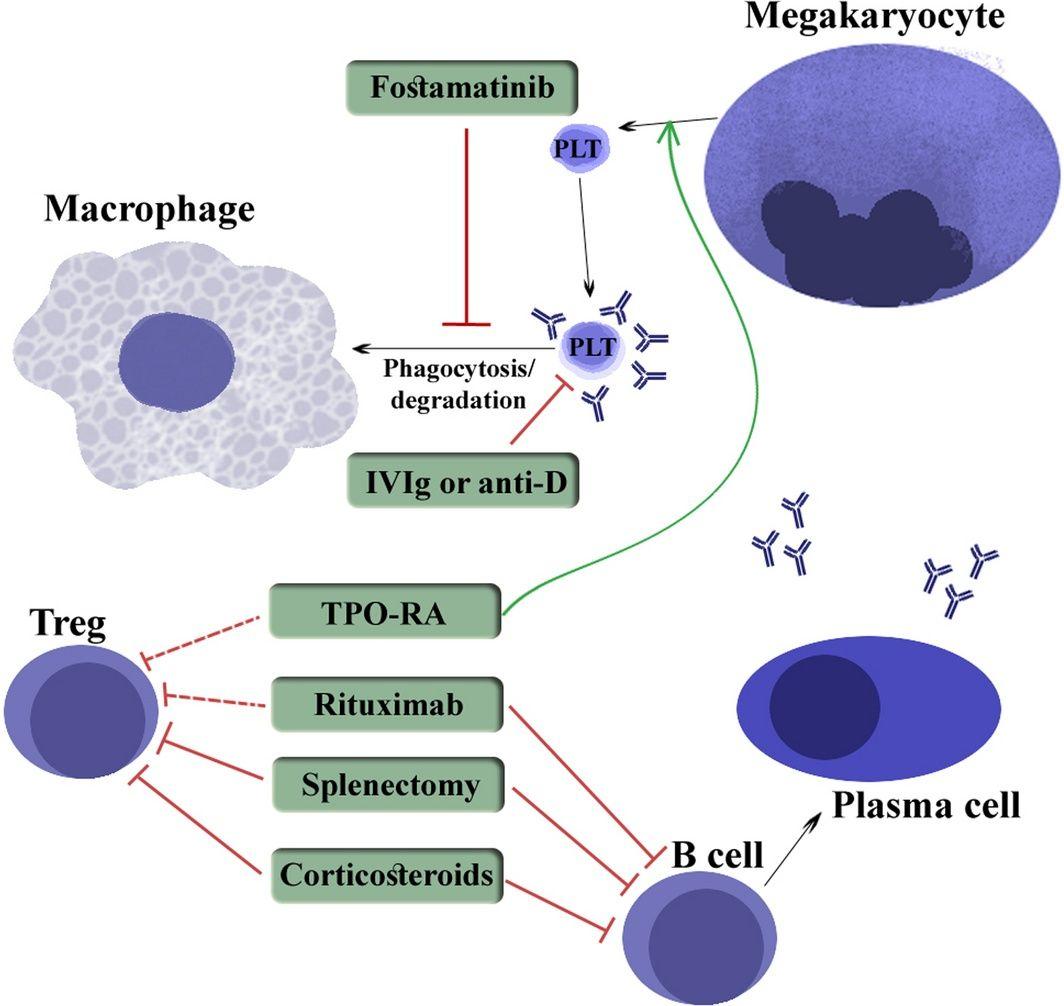
The following figure illustrates the mechanism of action of ITP therapies:
Active serious bleeding (emergency)
Per the McMaster ITP Emergency Management Guideline (2026), a critical bleed warrants combined use of: [10]
- Platelet transfusions (transient effect, use with other agents)
- High-dose corticosteroids (methylprednisolone 1 g IV or dexamethasone 40 mg IV)
- IVIG 1 g/kg (repeat day 2 if needed)
- Tranexamic acid (1 g IV or 1.3 g PO TID)
- TPO-receptor agonist (romiplostim or eltrombopag)
- Discontinue all anticoagulants and antiplatelet agents [1]
- Conditional recommendation for urgent splenectomy if all else fails; conditional recommendation against recombinant factor VIIa (thrombosis risk) [10]
Non-bleeding / minor bleeding
- Platelets ≥30 × 10³/μL, asymptomatic or minor bleeding: Observation with close follow-up [8]
- Platelets <30 × 10³/μL: Corticosteroids — prednisone 1–2 mg/kg/day × 1–2 weeks then taper, OR dexamethasone 40 mg × 4 days [1]
- Add IVIG if rapid platelet rise needed (pre-procedure, severe thrombocytopenia <10 × 10³/μL) [1]
Refractory/relapsed ITP
- TPO-receptor agonists (romiplostim, eltrombopag, avatrombopag) — preferred for sustained response [1][11]
- Rituximab — alternative; slower onset but durable in some patients [1]
- Fostamatinib — for chronic ITP after insufficient response to prior treatment [11]
- Splenectomy — defer until >12 months from diagnosis; reserved for medical treatment failure [1][6]
17. Disposition
Admission criteria (per ASH 2019 Guidelines): [8][12]
- Newly diagnosed ITP with platelets <20 × 10³/μL — admission suggested even if asymptomatic or with minor mucocutaneous bleeding
- Active significant bleeding at any platelet count
- Diagnostic uncertainty
- Significant comorbidities increasing bleeding risk
- Social concerns or inability to ensure follow-up
Outpatient management appropriate for: [8][12]
- Platelets ≥20 × 10³/μL with no or minor bleeding
- Established ITP (known diagnosis) with platelets <20 × 10³/μL if asymptomatic and previously responsive to rescue agents
- Reliable follow-up with hematology within 24–72 hours ensured [12]
Observation (no treatment)
Hematology consultation
- All newly diagnosed ITP patients should have hematology follow-up within 24–72 hours [12]
- Urgent consultation for refractory disease, diagnostic uncertainty, or consideration of splenectomy
18. Follow-Up / Return Precautions
Follow-up timing
- Hematology follow-up within 24–72 hours of new diagnosis or relapse [12]
- Repeat CBC within 1–2 weeks after initiating treatment to assess response
- If on corticosteroids: monitor glucose, blood pressure, and taper schedule
- If on TPO-RAs: regular CBC monitoring; watch for rebound thrombocytopenia on discontinuation [1]
- If on eltrombopag: LFTs at baseline and regularly during treatment [1]
Return precautions — instruct patients to seek immediate care for:
- New or worsening bleeding (nosebleeds not stopping with pressure, blood in urine/stool, heavy menstrual bleeding)
- Severe headache, vision changes, confusion, or weakness — concern for intracranial hemorrhage [4]
- Oral blood blisters (wet purpura)
- Dizziness, lightheadedness, or syncope (hemodynamic compromise)
- New bruising significantly worse than baseline
Patient counseling
- Avoid contact sports and activities with high injury risk while severely thrombocytopenic
- Avoid aspirin, NSAIDs, and other platelet-impairing medications
- Avoid alcohol
- Wear a medical alert bracelet if chronically thrombocytopenic
- In children, most cases resolve spontaneously within weeks to months; in adults, chronic disease is common but manageable [1][4][15]
References
1. Immune Thrombocytopenia. — Cooper N, Ghanima W. The New England Journal of Medicine. 2019.
2. Hematologic Emergencies: Recognition and Initial Management. — Jones DE, Walker JJ, Abellada AMP. American Family Physician. 2024.
3. Efficacy and Safety of Treatments in Newly Diagnosed Adult Primary Immune Thrombocytopenia: A Systematic Review and Network Meta-Analysis. — Wang Y, Sheng L, Han F, et al. EClinicalMedicine. 2023.
4. Immune thrombocytopenia. — National Library of Medicine (MedlinePlus) 2017.
5. Early Diagnosis and Tailored Treatment in Atypical Idiopathic Thrombocytopenic Purpura: A CARE Compliant Case Report. — Patel T, Kharat M, John JD, et al. Medicine. 2025.
6. Thrombocytopenia: Evaluation and Management. — Gauer RL, Whitaker DJ. American Family Physician. 2022.
7. Drug-Induced Immune Thrombocytopenia. — Aster RH, Bougie DW. The New England Journal of Medicine. 2007.
8. Updated Guidelines for Immune Thrombocytopenic Purpura: Expanded Management Options. — DeSouza S, Angelini D. Cleveland Clinic Journal of Medicine. 2021.
9. Immune Thrombotic Thrombocytopenic Purpura. — Pishko AM, Li A, Cuker A. The Journal of the American Medical Association. 2025.
10. Guideline on the Emergency Management of Critical Bleeding in Patients With Immune Thrombocytopenia. — Chowdhury SR, Sirotich E, Guyatt GH, et al. Blood Advances. 2026.
11. FDA Orange Book. — FDA Orange Book. 2026.
12. American Society of Hematology 2019 Guidelines for Immune Thrombocytopenia. — Neunert C, Terrell DR, Arnold DM, et al. Blood Advances. 2019.
13. ACOG Practice Bulletin No. 207: Thrombocytopenia in Pregnancy. — Committee on Practice Bulletins—Obstetrics Obstetrics and Gynecology. 2019.
14. Reference Guide for the Diagnosis of Adult Primary Immune Thrombocytopenia, 2023 Edition. — Kashiwagi H, Kuwana M, Murata M, et al. International Journal of Hematology. 2024.
15. Eltrombopag for Newly Diagnosed Pediatric Immune Thrombocytopenia Requiring Treatment. — Shimano KA, Grimes AB, Kaicker S, et al. The Journal of the American Medical Association. 2025.
16. Autoimmune thrombocytopenia: Current treatment options in adults with a focus on novel drugs. — Witkowski M, Witkowska M, Robak T. European Journal of Haematology. 2019.