Thrombotic Thrombocytopenic Purpura (TTP)
Immune TTP (iTTP) is a life-threatening thrombotic microangiopathy caused by autoantibodies against ADAMTS13, resulting in accumulation of ultra-large von Willebrand factor multimers, platelet-rich…
Immune TTP (iTTP) is a life-threatening thrombotic microangiopathy caused by autoantibodies against ADAMTS13, resulting in accumulation of ultra-large von Willebrand factor multimers, platelet-rich microthrombi, and ischemic organ injury. Untreated mortality approaches 90%; with modern triple therapy (therapeutic plasma exchange + corticosteroids + rituximab ± caplacizumab), 30-day survival exceeds 93%.[1-2] Annual incidence is 2–6 cases per million, with higher rates in females (IRR 3.19), Black individuals (IRR 7.09), and adults.[1]
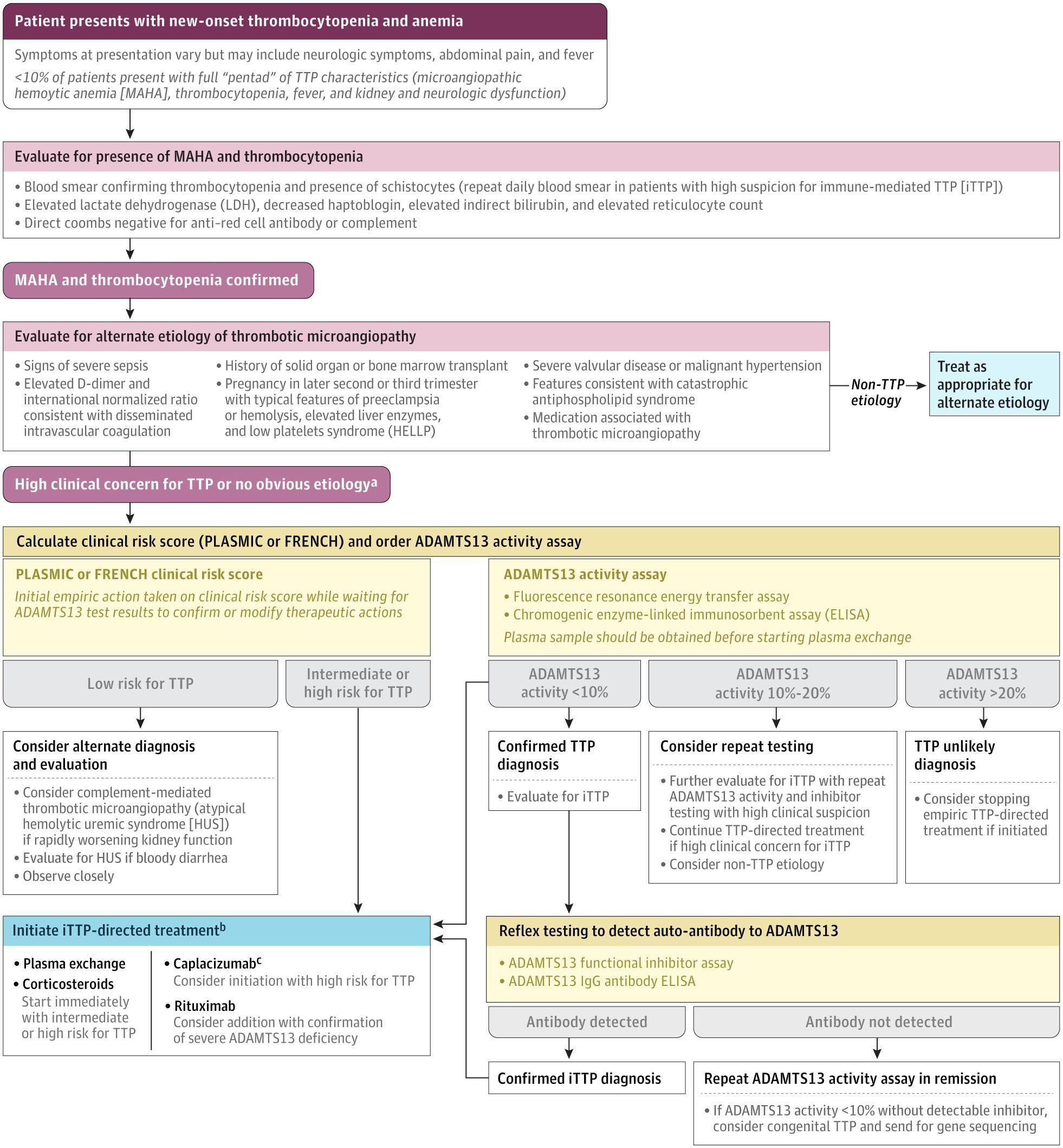
The following figure illustrates the diagnostic and initial management workflow:
View full figure Figure 2. Initial Diagnostic Workflow for Patients With Suspected Thrombotic Thrombocytopenic Purpura Immune Thrombotic Thrombocytopenic Purpura. JAMA. August 11, 2025.
1. History
- Key HPI questions: Onset and progression of fatigue, bruising/petechiae, headache, confusion, visual changes, chest pain, abdominal pain, dark urine, fever
- Symptom characterization: Neurologic symptoms are the most common presenting feature (39–80%), including headache, confusion, vision changes, and seizures; abdominal pain/nausea in 35–39%; fever in 10–35%[1]
- Timing/triggers: Acute onset over days; may be triggered by infection (HIV in 13%), pregnancy (7%), autoimmune disease (SLE, antiphospholipid syndrome in 14%), or rarely drugs (1%)[1]
- Important negatives: Absence of bloody diarrhea (argues against Shiga toxin HUS), absence of recent transplant or active malignancy, no recent heparin exposure
2. Alarm Features
- Neurologic deterioration: Seizures, focal deficits, stupor, coma — independent predictor of 30-day mortality[1]
- Cardiac involvement: Troponin elevation (present in 54–68% of patients), chest pain, heart failure, cardiogenic shock — elevated troponin above ULN associated with 6-fold increase in mortality (12.1% vs 2.0%)[3-4]
- Glasgow Coma Scale ≤14: 9-fold increase in mortality (20% vs 2.2%)[3]
- LDH >10× ULN: Independent predictor of 30-day mortality[1]
- Refractory disease: Lack of platelet count doubling after ≥5 TPE sessions with persistently elevated LDH[1]
3. Medications
Relevant medication contributors (drug-induced TMA)
- Definite causal association: quinine (most common), cyclosporine, tacrolimus, ticlopidine, gemcitabine, mitomycin[1][5]
- Other reported associations: clopidogrel, oxaliplatin, interferon-β, quetiapine, vancomycin[6-7]
Core treatments
- Therapeutic plasma exchange (TPE): Daily, 1–1.5 plasma volumes
- Corticosteroids: Prednisone 1 mg/kg/day PO or methylprednisolone 1000 mg IV daily × 3 days for severe features[1]
- Rituximab: 375 mg/m² weekly × 4 doses (off-label for TTP)[1]
- Caplacizumab: 11 mg IV bolus before first TPE, then 11 mg SC daily for ≥30 days after TPE cessation[8]
Contraindicated medications
- Platelet transfusions are generally avoided unless life-threatening hemorrhage (theoretical concern for fueling microvascular thrombosis)[9]
- Avoid re-exposure to any suspected causative drug
4. Diet
- No specific dietary triggers for iTTP
- Ensure adequate hydration, particularly during TPE
- Monitor for electrolyte derangements during plasma exchange (hypocalcemia from citrate anticoagulant)
5. Review of Systems
- Neurologic: Headache, confusion, vision changes, seizures, focal weakness, speech difficulty
- Cardiac: Chest pain, dyspnea, palpitations, syncope
- GI: Abdominal pain, nausea, vomiting
- Hematologic: Easy bruising, petechiae, mucosal bleeding, dark urine (hemoglobinuria)
- Renal: Decreased urine output (though severe renal failure is uncommon — only 5% develop kidney failure)[1]
- Constitutional: Fever, fatigue, malaise
6. Collateral History and Family History
- Prior episodes of TTP or unexplained thrombocytopenia/anemia (relapse occurs in ~30% at 5-year follow-up)[1]
- Family history of TTP (rare congenital TTP from biallelic ADAMTS13 mutations — 3–5% of cases)[1]
- History of autoimmune disease (SLE, antiphospholipid syndrome)
- HIV status, pregnancy status
- Medication history with focus on quinine, calcineurin inhibitors, ticlopidine, gemcitabine
7. Risk Factors
- Female sex (IRR 3.19)[1]
- Black race (IRR 7.09)[1]
- Adult age (peak incidence in 30s–50s)[1]
- Autoimmune disease (SLE, antiphospholipid syndrome — 14%)[1]
- HIV infection (13%)[1]
- Pregnancy (7%, particularly second/third trimester)[1]
- Obesity — associated with higher relapse risk in some cohorts
- Prior TTP episode — 16% clinical relapse rate, 13% ADAMTS13 relapse only at 5 years[1]
8. Differential Diagnosis
- Export Diagnosis Key Distinguishing Features References Complement-mediated TMA (aHUS) Platelets typically >30 × 10⁹/L, creatinine >2 mg/dL, complement dysregulation[1]
- Shiga toxin HUS Bloody diarrhea prodrome, E. coli O157:H7, more common in children[1]
- DIC Prolonged PT/aPTT, low fibrinogen, elevated D-dimer, underlying sepsis/malignancy[1]
- HELLP syndrome Pregnancy (2nd/3rd trimester), elevated liver enzymes, resolves with delivery[1]
- Severe vitamin B12 deficiency Elevated MCV (mean 109 fL), very high LDH (mean 3539 U/L), low reticulocyte count, near-normal bilirubin[1]
- Catastrophic antiphospholipid syndrome Antiphospholipid antibodies, multi-organ thrombosis, often in SLE[1]
- Malignant hypertension Severe hypertension, retinopathy, renal injury[1]
- Evans syndrome Positive direct Coombs test (DAT), autoimmune hemolysis + ITP[2]
- Drug-induced TMA Temporal association with offending drug (quinine, cyclosporine, gemcitabine); ADAMTS13 typically >10%[3-4]
9. Past Medical History
- Prior TTP episodes (relapse risk is significant — 40% at 5-year follow-up in UK registry)[12]
- Autoimmune conditions (SLE, antiphospholipid syndrome, thyroid disease)
- HIV/AIDS
- Prior pregnancy-related TMA
- History of splenectomy
- Transplant history (solid organ or stem cell)
- Active malignancy
10. Physical Exam
- Vitals: Fever (10–35%), tachycardia, hypo- or hypertension
- Skin: Petechiae, purpura, ecchymoses, jaundice/scleral icterus
- Neurologic: Altered mental status, confusion, focal deficits, seizures, visual field deficits — perform thorough neurologic exam including GCS
- Cardiac: Tachycardia, signs of heart failure (JVD, crackles, S3), arrhythmias
- Abdominal: Tenderness (often diffuse), hepatosplenomegaly (uncommon)
- Note: Less than 10% present with the classic "pentad" (hemolytic anemia, thrombocytopenia, fever, renal dysfunction, neurologic dysfunction)[1]
11. Lab Studies
Diagnostic labs
- CBC with peripheral smear: Thrombocytopenia (typically <30 × 10⁹/L), anemia, schistocytes ≥1% (may be absent initially — repeat daily)[1]
- ADAMTS13 activity: <10% confirms diagnosis; draw before initiating TPE if possible[1]
- Anti-ADAMTS13 antibody (IgG ELISA and/or functional inhibitor assay)[1]
- Hemolysis panel: LDH (markedly elevated), indirect bilirubin (elevated), haptoglobin (undetectable), reticulocyte count (elevated)
- Direct Coombs test (DAT): Negative (distinguishes from autoimmune hemolytic anemia)
- Coagulation studies: PT/INR and aPTT typically normal or mildly prolonged (helps distinguish from DIC)[1]
- BMP: Creatinine typically <2 mg/dL (distinguishes from aHUS)
- Troponin: Elevated in 54–68%; prognostic marker — cTnI >0.25 μg/L associated with 3-fold increase in death/refractoriness[4][13]
Rule-out labs
- Blood cultures (if febrile, to exclude sepsis)
- HIV testing, hepatitis B panel (before rituximab)
- ANA, antiphospholipid antibodies (if autoimmune etiology suspected)
- Pregnancy test in women of childbearing age
- Stool studies if diarrhea present (Shiga toxin)
12. Imaging
- CT head without contrast: If focal neurologic deficits or seizures — evaluate for stroke, hemorrhage
- MRI brain: More sensitive for ischemic changes from microvascular thrombosis; consider if CT is negative with persistent neurologic symptoms
- Echocardiogram: If troponin elevated or signs of heart failure — assess for wall motion abnormalities, reduced EF, pericardial effusion[14-15]
- Chest X-ray: If respiratory symptoms or heart failure suspected
- Routine imaging is not required for diagnosis
13. Special Tests
- PLASMIC Score (most commonly used clinical prediction tool):[1]
- Export Parameter Points References Platelet count 30 × 10⁹/L +1[1]
- Combined hemoLysis variable (indirect bilirubin >2 mg/dL, reticulocytes >2.5%, or undetectable haptoglobin) +1[1]
- Absence of active cancer +1[1]
- Absence of Stem-cell or solid-organ transplant +1[1]
- MCV 90 fL +1[1]
- INR 1.5 +1[1]
- Creatinine 2 mg/dL +1[1]
- Score 0–4: Low risk → TTP unlikely
- Score 5: Intermediate risk → consider empiric treatment, urgent hematology consult
- Score 6–7: High risk → initiate empiric treatment immediately
- Sensitivity of score ≥5: 99% (95% CI, 91–100%); specificity: 57%[1][16]
- Rapid ADAMTS13 assays (e.g., HemosIL AcuStar): Turnaround <1 hour, sensitivity 98%, specificity 99% — may avert unnecessary empiric TPE in non-TTP patients[17]
14. ECG
- Obtain ECG on all patients with suspected TTP
- Findings may include: ST-segment changes (depression or elevation), T-wave inversions, repolarization abnormalities — present in 62% of patients with elevated troponin[13]
- Non-ST elevation pattern is the predominant presentation of myocardial injury in TTP[15]
- Conduction abnormalities: AV block (microthrombi in AV node and His bundle on autopsy)[18]
- Arrhythmias: Atrial tachyarrhythmias (5.9%), ventricular tachyarrhythmias (2.0%)[14]
- Cardiac arrest reported in 1.9% of TTP hospitalizations[14]
15. Assessment
- iTTP is a hematologic emergency — mortality approaches 90% without treatment[2]
- The classic pentad (MAHA, thrombocytopenia, fever, neurologic dysfunction, renal dysfunction) is present in <10% of cases; do not wait for the full pentad to initiate treatment[1]
- Severity stratification based on: neurologic features, troponin elevation, GCS, LDH level, ADAMTS13 antibody burden[1][3][19]
- Cardiovascular complications occur in 25% of hospitalizations and are associated with significantly higher in-hospital mortality (19.7% vs 4.1%)[14]
- Long-term survivors have elevated cardiovascular mortality and a 5-fold higher rate of ischemic stroke compared to the general population[1]
16. Treatment Plan
Initial stabilization
- Emergent hematology consultation
- Large-bore IV access; central venous catheter for apheresis
First-line therapy (ISTH 2020 guidelines, reaffirmed 2025)
- Therapeutic plasma exchange (TPE): Daily, 1–1.5 plasma volumes with FFP replacement. Continue until sustained platelet normalization (≥150 × 10⁹/L for ≥2 days) and declining LDH
- Corticosteroids: Prednisone 1 mg/kg/day PO or methylprednisolone 1 g IV daily × 3 days for severe neurologic/cardiac features[1]
- Rituximab: 375 mg/m² IV weekly × 4 doses (conditional recommendation). Screen for hepatitis B before initiation[1]
- Caplacizumab: 11 mg IV bolus before first TPE, then 11 mg SC daily. Continue for 30 days after last TPE or until ADAMTS13 recovery >10%. May extend up to additional 28 days if ADAMTS13 remains severely deficient[1][8]
Key outcomes with triple/quadruple therapy
- Caplacizumab reduces time to platelet normalization (median ~2.7 vs 3.5 days), decreases exacerbations (RD −29%), and reduces refractory disease (RD −8%), but increases bleeding risk (any bleeding 58.5%)[1]
Refractory TTP (no response after ≥5 TPE sessions)
- Escalate to twice-daily TPE
- Add rituximab and/or caplacizumab if not already initiated
- Consider cyclosporine, cyclophosphamide, vincristine, bortezomib, daratumumab, or splenectomy[1]
17. Disposition
- All patients with suspected TTP require hospital admission, typically to the ICU or a monitored setting with apheresis capability[21]
- Transfer to a center with TPE capability if not available locally
- Admission criteria: Any patient with MAHA + thrombocytopenia and PLASMIC ≥5 or high clinical suspicion
- Discharge criteria: Sustained platelet count normalization (≥150 × 10⁹/L for ≥2 consecutive days), declining LDH, no new organ injury, transition to outpatient caplacizumab (if applicable)
- Hematology consultation is mandatory and should be obtained emergently[1]
18. Follow Up / Return Precautions
Monitoring in remission
- ADAMTS13 activity should be monitored regularly (e.g., every 1–3 months initially, then every 3–6 months) — declining ADAMTS13 <20% warrants preemptive rituximab[1]
- Preemptive rituximab for ADAMTS13 relapse (activity <10–20% without clinical symptoms) reduces clinical relapse from 0.5 episodes/year to 0 episodes/year (OR 0.09)[1][22]
- CBC, LDH, haptoglobin at follow-up visits
- Cardiovascular risk assessment: Survivors have elevated long-term cardiovascular mortality; ischemic stroke occurs in 13% during remission (5-fold higher than expected)[1]
- Return precautions — instruct patients to seek immediate care for:
- New or worsening headache, confusion, vision changes, weakness
- Chest pain, shortness of breath, palpitations
- New bruising, petechiae, dark urine
- Fever
Expected course
- 16% of patients experience at least one clinical relapse; ~30% have clinical or ADAMTS13 relapse at 5 years[1]
- Long-term follow-up with hematology is essential for relapse surveillance, cardiovascular risk management, and psychosocial support[1][23]
References
1. Immune Thrombotic Thrombocytopenic Purpura. — Pishko AM, Li A, Cuker A. The Journal of the American Medical Association. 2025.
2. Bleeding and Coagulopathies in Critical Care. — Hunt BJ. The New England Journal of Medicine. 2014.
3. Presenting ADAMTS13 Antibody and Antigen Levels Predict Prognosis in Immune-Mediated Thrombotic Thrombocytopenic Purpura. — Alwan F, Vendramin C, Vanhoorelbeke K, et al. Blood. 2017.
4. Cardiac Troponin-I on Diagnosis Predicts Early Death and Refractoriness in Acquired Thrombotic Thrombocytopenic Purpura. Experience of the French Thrombotic Microangiopathies Reference Center. — Benhamou Y, Boelle PY, Baudin B, et al. Journal of Thrombosis and Haemostasis : JTH. 2015.
5. Drug-Induced Thrombotic Microangiopathy: A Systematic Review of Published Reports. — Al-Nouri ZL, Reese JA, Terrell DR, Vesely SK, George JN. Blood. 2015.
6. Syndromes of Thrombotic Microangiopathy. — George JN, Nester CM. The New England Journal of Medicine. 2014.
7. D rug‐induced thrombotic microangiopathy: E xperience of the O klahoma registry and the BloodCenter of W isconsin. — Reese JA, Bougie DW, Curtis BR, et al. American Journal of Hematology. 2015.
8. FDA Drug Label. — Updated date: 2026-04-24. Food and Drug Administration.
9. Hematologic Emergencies: Recognition and Initial Management. — Jones DE, Walker JJ, Abellada AMP. American Family Physician. 2024.
10. Thrombotic thrombocytopenic purpura masquerading as Evans syndrome. — Mudra SE, Ardoin K, Aggarwal V, et al. American Journal of Hematology. 2024.
11. Thrombocytopenia: Evaluation and Management. — Gauer RL, Whitaker DJ. American Family Physician. 2022.
12. Long-Term Risk of Relapse in Immune-Mediated Thrombotic Thrombocytopenic Purpura and the Role of Anti-Cd20 Therapy. — Doyle AJ, Stubbs MJ, Dutt T, et al. Blood. 2023.
13. Cardiac Involvement in Acute Thrombotic Thrombocytopenic Purpura: Association With Troponin T and IgG Antibodies to ADAMTS 13. — Hughes C, McEwan JR, Longair I, et al. Journal of Thrombosis and Haemostasis : JTH. 2009.
14. Cardiovascular Complications and Their Association With Mortality in Patients With Thrombotic Thrombocytopenic Purpura. — Balasubramaniyam N, Yandrapalli S, Kolte D, et al. The American Journal of Medicine. 2021.
15. Myocardial necrosis in patients with thrombotic thrombocytopenic purpura: pathophysiology and rationale for specific therapy. — Sane DC, Streer NP, Owen J. European Journal of Haematology. 2009.
16. Diagnostic Accuracy of the PLASMIC Score in Patients With Suspected Thrombotic Thrombocytopenic Purpura: A Systematic Review and Meta-Analysis. — Paydary K, Banwell E, Tong J, Chen Y, Cuker A. Transfusion. 2020.
17. Rapid ADAMTS13 Activity Assays for Thrombotic Thrombocytopenic Purpura: A Systematic Review and Meta-Analysis. — Deshpande SR, Tarawneh H, Deitelzweig C, et al. Blood. 2025.
18. The Heart and Cardiac Conduction System in Thrombotic Thrombocytopenic Purpura. A Clinicopathologic Study of 17 Autopsied Patients. — Ridolfi RL, Hutchins GM, Bell WR. Annals of Internal Medicine. 1979.
19. Prognostic Value of Laboratory Biomarkers for Mortality Risk Stratification in Thrombotic Thrombocytopenic Purpura. — Xiang X, Dai YQ. Annals of Hematology. 2025.
20. 2025 Focused Update of the 2020 ISTH Guidelines for Management of Thrombotic Thrombocytopenic Purpura. — Zheng XL, Al-Housni Z, Cataland SR, et al. Journal of Thrombosis and Haemostasis : JTH. 2025.
21. The Standard of Care for Immune Thrombotic Thrombocytopenic Purpura Today. — Zheng XL. Journal of Thrombosis and Haemostasis : JTH. 2021.
22. Preemptive Rituximab Infusions After Remission Efficiently Prevent Relapses in Acquired Thrombotic Thrombocytopenic Purpura. — Hie M, Gay J, Galicier L, et al. Blood. 2014.
23. Thrombotic Thrombocytopenic Purpura: Pathophysiology, Diagnosis, and Management. — Sukumar S, Lämmle B, Cataland SR. Journal of Clinical Medicine. 2021.