Transverse Myelitis
Transverse myelitis (TM) is an inflammatory disorder of the spinal cord characterized by acute onset of motor, sensory, and autonomic dysfunction, evolving over hours to days.[1-2] It may be idiopa…
Transverse myelitis (TM) is an inflammatory disorder of the spinal cord characterized by acute onset of motor, sensory, and autonomic dysfunction, evolving over hours to days.[1-2] It may be idiopathic or associated with demyelinating diseases (MS, NMOSD, MOGAD), autoimmune conditions (SLE, Sjögren's), or infections.[3-4] Incidence is approximately 1–8 per million per year, with ~20% of cases occurring in children.[5]
1. History
- Onset and tempo: Acute to subacute progression over hours to days; symptoms reaching nadir typically within 4 hours to 21 days[1][6]
- Motor: Bilateral weakness (paraparesis or quadriparesis depending on level); may be asymmetric[1]
- Sensory: Band-like tightness, numbness, or paresthesias; neuropathic pain (midline aching or dermatomal/lancinating); Lhermitte's sign[1]
- Autonomic: Urinary retention or incontinence, bowel dysfunction, sexual dysfunction[1]
- Antecedent events: Preceding viral illness or vaccination in up to 66% of cases (1–4 weeks prior)[1][7]
- Important negatives: Absence of trauma, recent back surgery, known malignancy, IV drug use, immunosuppression
2. Alarm Features
- Respiratory compromise: High cervical cord involvement (C3–C5) threatening diaphragmatic function[5][8]
- Rapidly ascending weakness mimicking Guillain-Barré syndrome[1]
- Spinal shock: Severe weakness + hypotonia + areflexia — the only recognized predictor of poor outcome[1]
- Complete transverse myelitis (bilateral motor/sensory/autonomic loss) — associated with worse prognosis and higher relapse risk[3][9]
- Hyperacute onset (minutes) — consider spinal cord infarction rather than TM[10-11]
- Progressive course beyond 21 days — suggests alternative etiology (dural AV fistula, tumor, metabolic)[6][10]
3. Medications
- First-line treatment: IV methylprednisolone 1 g daily for 3–5 days[1][12]
- Second-line (steroid-refractory): Therapeutic plasma exchange (PLEX), 5–7 sessions exchanging 1.1–1.5 plasma volumes each[1][8]
- Third-line: IV immunoglobulin (IVIg) 2 g/kg over 2–5 days; cyclophosphamide in select cases (e.g., SLE-associated TM)[8][13]
- Contraindicated: Avoid immunosuppressives until infectious etiologies are reasonably excluded; however, empiric corticosteroids should not be delayed as they generally do not worsen infectious or vascular mimics[8]
- Symptomatic: Gabapentin/pregabalin for neuropathic pain; baclofen or tizanidine for spasticity; anticholinergics or intermittent catheterization for bladder dysfunction
4. Diet
- No specific dietary triggers for TM
- Adequate hydration is important, particularly with autonomic dysfunction and neurogenic bladder
- High-fiber diet to manage neurogenic bowel/constipation
- Long-term: Nutritional optimization including vitamin D supplementation if deficient (relevant if MS is the underlying etiology)
5. Review of Systems
- Neurologic: Visual changes (optic neuritis → MS/NMOSD), encephalopathy (ADEM), seizures
- Ophthalmologic: Eye pain with movement, color desaturation (optic neuritis)
- Rheumatologic: Joint pain, rash, oral ulcers, dry eyes/mouth (SLE, Sjögren's, Behçet's)
- Infectious: Fever, rash, recent travel, tick exposure, HIV risk factors
- Constitutional: Weight loss, night sweats (malignancy, sarcoidosis, TB)
- Respiratory: Cough, dyspnea (sarcoidosis, respiratory failure from high cord lesion)
6. Collateral History and Family History
- Prior episodes of neurologic symptoms (optic neuritis, limb weakness) suggesting relapsing disease
- Family history of autoimmune disease (MS, SLE, NMOSD)
- Vaccination history (recent immunization as potential trigger)[4]
- Social history: HIV risk factors, IV drug use, travel to endemic areas (schistosomiasis, HTLV-1)
7. Risk Factors
- Infectious prodrome within 1 month (most common association)[7]
- Autoimmune disease: SLE, Sjögren's, sarcoidosis, antiphospholipid syndrome[3][14]
- Demyelinating disease: MS, NMOSD, MOGAD[3]
- Age: Bimodal distribution — peaks at 10–19 and 30–39 years[5]
- Female sex: Higher risk for NMOSD-associated TM
- COVID-19 infection or vaccination has been associated with TM in case reports[4]
8. Differential Diagnosis
Cannot-miss diagnoses
- Spinal cord compression (epidural abscess, hematoma, tumor) — requires emergent surgical evaluation
- Spinal cord infarction — hyperacute onset (minutes), anterior cord syndrome, owl-eye/snake-eye pattern on axial MRI[10-11]
- Cauda equina syndrome — LMN signs, saddle anesthesia
Key differentials
- Multiple sclerosis — short-segment, peripheral, asymmetric lesions; brain lesions typical of MS; OCBs in 80–90%[12][15]
- NMOSD (AQP4+) — longitudinally extensive (≥3 segments), central lesions, bright spotty T2 lesions, AQP4-IgG positive; worse prognosis[11][15]
- MOGAD — longitudinally extensive, central gray matter "H-sign," MOG-IgG positive; generally better motor outcomes than NMOSD[3][16]
- Spinal dural arteriovenous fistula — older males, progressive/fluctuating course, lower thoracic predominance, flow voids on MRI[10][17]
- Neurosarcoidosis — dorsal subpial enhancement, trident sign, positive body PET/CT[10-11]
- Metabolic myelopathy — B12/copper deficiency, dorsal column involvement, inverted V-sign[10-11]
- Infectious myelitis — VZV, HIV, HTLV-1, enterovirus, syphilis[14]
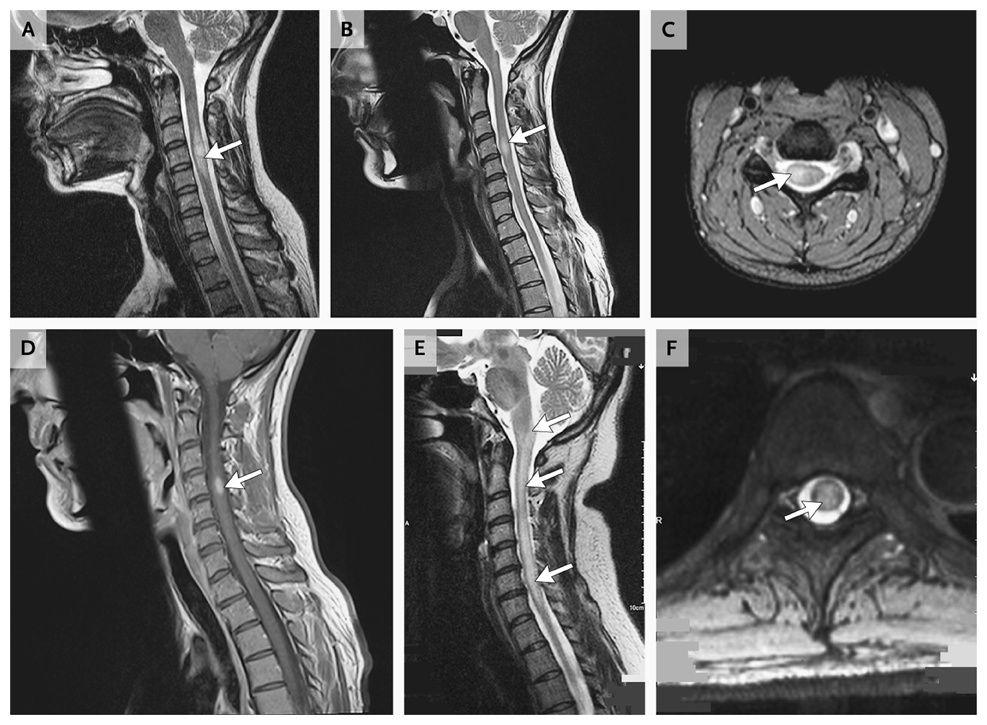
- The following figure from the NEJM illustrates key MRI differences between idiopathic TM, MS-associated myelitis, and NMO-associated myelitis:
9. Past Medical History
- Prior episodes of optic neuritis, myelitis, or brainstem syndromes (relapsing demyelinating disease)
- Known autoimmune conditions (SLE, Sjögren's, sarcoidosis)
- History of malignancy (paraneoplastic myelopathy)
- Immunosuppression or HIV status
- Surgical history (prior spinal procedures, aortic surgery → cord ischemia risk)
10. Physical Exam
- Vital signs: Monitor respiratory rate and oxygen saturation closely (high cervical lesions)
- Motor: Paraparesis or quadriparesis; initially may have flaccid tone (spinal shock) → later spasticity with UMN signs[1]
- Reflexes: Hyperreflexia and Babinski sign (confirms central/UMN lesion); areflexia in spinal shock[1]
- Sensory: Well-defined truncal sensory level — below which pain/temperature sensation is altered; distinguishes myelopathy from cerebral or peripheral causes[1]
- Lhermitte's sign: Paresthesias radiating down spine/limbs with neck flexion[1]
- Autonomic: Distended bladder (retention), decreased rectal tone
- Paroxysmal tonic spasms: Involuntary dystonic contractions of limb/trunk muscles[1]
- Fundoscopy: Optic disc pallor or papillitis if concurrent optic neuritis
11. Lab Studies
- Serum: CBC, CMP, ESR/CRP, ANA, dsDNA, SSA/SSB, ACE level, vitamin B12, copper, folate, HIV, RPR/VDRL, HTLV-1/2 antibodies[14][18]
- Antibody testing: AQP4-IgG (NMO-IgG) and MOG-IgG (cell-based assay preferred) — critical for determining etiology and relapse risk[3][12]
CSF analysis
- Cell count and differential (pleocytosis in ~50%; ≥50 WBC/mm³ suggests NMO)
- Protein (elevated in ~48%)
- Glucose
- Oligoclonal bands and IgG index (OCBs present in 80–90% of MS vs ~23% of NMO)
- Cytology and flow cytometry (rule out lymphoma/carcinomatous meningitis)
- Infectious studies: VZV PCR/serology, HSV PCR, enterovirus PCR, Gram stain, bacterial/fungal cultures, syphilis, Lyme
12. Imaging
- First-line: MRI cervical and thoracic spine without and with IV contrast — the ACR rates this as "usually appropriate" for suspected TM[3]
- T2 hyperintensity with cord swelling is the hallmark finding[3]
- Gadolinium enhancement detects active inflammation (common but not universal)[3]
- Image the entire spine to assess lesion extent and exclude compressive pathology[3]
- Brain MRI with contrast: Essential to evaluate for MS-type lesions (periventricular, juxtacortical, infratentorial) or NMOSD-pattern brain lesions[12]
- CT spine: Less sensitive than MRI; consider only if MRI is contraindicated[3]
- CT myelography: Alternative when cord compression is suspected and MRI is unavailable[12]
Key MRI patterns
- MS: Short-segment (<3 segments), peripheral, asymmetric
- NMOSD: Longitudinally extensive (≥3 segments), central, bright spotty lesions
- MOGAD: Longitudinally extensive, central gray matter H-sign, fluffy/poorly demarcated
- Idiopathic TM: Central hyperintensity spanning 3–4 segments with focal peripheral enhancement
13. Special Tests
- ASIA (American Spinal Injury Association) Impairment Scale: Standardized severity grading for spinal cord dysfunction[8]
- Modified Rankin Scale (mRS): Functional outcome assessment[20]
- Nerve conduction studies/EMG: If peripheral nervous system involvement is suspected (concurrent GBS, overlap syndromes); PNS involvement is an independent predictor of poor outcome[9]
- Visual evoked potentials: If subclinical optic neuritis is suspected (supports MS diagnosis)
- CT chest/body PET-CT: If sarcoidosis is suspected[11]
- Spinal angiography: Gold standard if dural AV fistula is suspected[17]
14. ECG
- ECG is not a primary diagnostic tool for TM
- Obtain ECG if autonomic dysfunction is present (bradycardia, arrhythmias from high cervical/thoracic cord lesions)
- Baseline ECG before initiating plasma exchange (catheter-related complications, electrolyte shifts)
- Rule out cardiac source of embolism if spinal cord infarction is in the differential
15. Assessment
- TM is a clinical syndrome — not a final diagnosis. The critical task is to determine the underlying etiology, as this dictates treatment, relapse risk, and prognosis.[1][3]
Severity stratification
- Partial TM: Asymmetric deficits, partial sensory/motor involvement — more likely MS-associated[3]
- Complete TM: Bilateral symmetric motor/sensory/autonomic loss — associated with NMOSD, idiopathic TM, or systemic autoimmune disease; higher disability risk[3][9]
Prognosis
- ~50–70% of patients achieve partial or complete recovery[1]
- Most recovery occurs within the first 3 months, though improvement may continue for ≥1 year[1]
- At 6 months, 60% have no or negligible symptoms (mRS 0), 29% have minor symptoms (mRS 1), and 11% have debilitating symptoms (mRS ≥2)[20]
- NMOSD-associated TM carries the worst prognosis: 38% with mRS ≥2 at 6 months vs 2% for MS-associated TM[20]
- Predictors of poor outcome: older age (≥50), complete TM, spinal shock, PNS involvement, elevated CSF PMNs, elevated albumin ratio[1][9][20]
16. Treatment Plan
Initial stabilization
- ABCs — monitor respiratory function closely for high cervical lesions; intubation if FVC declining
- Foley catheter for urinary retention
- DVT prophylaxis (pharmacologic + mechanical)
- Pain management (neuropathic pain agents)
Acute immunotherapy
- IV methylprednisolone 1 g/day × 3–5 days — first-line; should not be delayed while awaiting workup[1][8][12]
- If no improvement or severe presentation: Plasma exchange (PLEX) — 5–7 sessions over ~2 weeks; 42% of steroid-refractory patients showed moderate-to-marked improvement vs 6% with sham[1][12]
- IVIg (2 g/kg over 2–5 days) is an alternative second-line option, particularly in pediatric patients or when PLEX is not feasible[13]
- Oral steroid taper is controversial and not routinely required if good recovery after IV pulse[13]
Long-term disease-specific therapy
- NMOSD: Rituximab, eculizumab, satralizumab, or inebilizumab
- MS: Disease-modifying therapies per MS guidelines
- Sarcoidosis: Prolonged oral corticosteroids (prednisone 1 mg/kg/day for 6–12 months)
- Idiopathic TM: Observation with close follow-up; no established long-term therapy
Rehabilitation
- Early physical and occupational therapy consultation[1]
- Bladder management program
- Bowel regimen
- Psychological support
17. Disposition
- Admit all patients with suspected acute TM for observation, IV corticosteroids, and monitoring of neurologic progression[1]
- ICU admission: High cervical lesions with respiratory compromise, rapidly ascending weakness, hemodynamic instability from autonomic dysfunction
- Neurology consultation: All cases — for diagnostic workup, antibody testing, and treatment decisions
- Neurosurgery consultation: If compressive myelopathy cannot be excluded on initial imaging
- Inpatient rehabilitation: For patients with significant residual motor or functional deficits after acute treatment
- Discharge criteria: Neurologic stability or improvement, adequate bladder/bowel management, safe mobility, outpatient neurology follow-up arranged
18. Follow Up / Return Precautions
- Neurology follow-up within 1–2 weeks of discharge for antibody results review and treatment planning[12]
- Repeat MRI at 3–6 months to assess lesion evolution and screen for new lesions[12]
- Relapse monitoring: ~34% of non-MS/non-NMOSD patients experience ≥1 neuroinflammatory relapse; relapse rate is 5.9% per year[20]
- Risk factors for relapse: Presence of OCBs, transverse/multifocal spinal cord lesions on MRI[20]
- Return precautions — instruct patients to seek immediate care for:
- New or worsening weakness in any extremity
- New sensory changes or ascending numbness
- Loss of bladder or bowel control
- Difficulty breathing or swallowing
- New visual changes (eye pain, vision loss)
- Expected recovery: Most improvement occurs in the first 3 months; continued gains possible up to 1–2 years. Common long-term sequelae include sensory disturbances (15–50%), bladder dysfunction, and residual motor deficits.[1][8]
References
1. Transverse Myelitis. — Frohman EM, Wingerchuk DM. The New England Journal of Medicine. 2010.
2. Clinical Biomarkers Differentiate Myelitis From Vascular and Other Causes of Myelopathy. — Barreras P, Fitzgerald KC, Mealy MA, et al. Neurology. 2018.
3. ACR Appropriateness Criteria® Demyelinating Diseases. — Expert Panel on Neurologic Imaging, Kalnins A, Lewis LM, et al. Journal of the American College of Radiology : JACR. 2026.
4. Clinical Profile and Outcomes of COVID-19-Associated Transverse Myelitis: A Case Report and Review of Literature. — Gudlavalleti A, Nath A. Neurology. Clinical Practice. 2022.
5. Pediatric Acute Transverse Myelitis Overview and Differential Diagnosis. — Wolf VL, Lupo PJ, Lotze TE. Journal of Child Neurology. 2012.
6. Spinal Cord Involvement in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders. — Ciccarelli O, Cohen JA, Reingold SC, Weinshenker BG, et al. The Lancet. Neurology. 2019.
7. Safety and Efficacy of Plasma Exchange in Pediatric Transverse Myelitis. — Noland DK, Greenberg BM. Neurology. Clinical Practice. 2018.
8. Pediatric Transverse Myelitis. — Absoud M, Greenberg BM, Lim M, et al. Neurology. 2016.
9. Predictors of Outcome in a Large Retrospective Cohort of Patients With Transverse Myelitis. — Gastaldi M, Marchioni E, Banfi P, et al. Multiple Sclerosis. 2018.
10. Clinical Reasoning: A 57-Year-Old Man With Stepwise Progressive Paraparesis, Sensory Loss, Urinary Retention, and Constipation. — Alkabie S, Tanweer O, Hutton GJ, Cuascut FX. Neurology. 2022.
11. Identifying Specific Myelopathy Etiologies in the Evaluation of Suspected Myelitis: A Retrospective Analysis. — Alkabie S, Casserly CS, Morrow SA, Racosta JM. Journal of the Neurological Sciences. 2023.
12. Approach to Acute or Subacute Myelopathy. — Schmalstieg WF, Weinshenker BG. Neurology. 2010.
13. Paediatric Multiple Sclerosis and Antibody-Associated Demyelination: Clinical, Imaging, and Biological Considerations for Diagnosis and Care. — Fadda G, Armangue T, Hacohen Y, Chitnis T, Banwell B. The Lancet. Neurology. 2021.
14. Mystery Case: A 61-Year-Old Woman With Lower Extremity Paralysis and Sensory Loss. — Manners J, Jadhav AP, Xia Z. Neurology. 2017.
15. Differentiating Multiple Sclerosis From AQP4-Neuromyelitis Optica Spectrum Disorder and MOG-Antibody Disease With Imaging. — Cortese R, Prados Carrasco F, Tur C, et al. Neurology. 2023.
16. Comparison of Clinical Outcomes of Transverse Myelitis Among Adults With Myelin Oligodendrocyte Glycoprotein Antibody vs Aquaporin-4 Antibody Disease. — Mariano R, Messina S, Kumar K, et al. JAMA Network Open. 2019.
17. Clinical, Radiological, and CSF Features Distinguishing Spinal Dural Arteriovenous Fistula From Idiopathic Transverse Myelitis and Seropositive NMOSD-/MOGAD-associated Myelopathy: A Retrospective Observational Study. — Sarıdaş F, Özpar R, Ceylan D, et al. Scientific Reports. 2026.
18. Non-Icans Neurological Complications After CAR T-Cell Therapies: Recommendations From the EBMT Practice Harmonisation and Guidelines Committee. — Graham CE, Velasco R, Alarcon Tomas A, et al. The Lancet. Oncology. 2025.
19. Evaluation of Idiopathic Transverse Myelitis Revealing Specific Myelopathy Diagnoses. — Zalewski NL, Flanagan EP, Keegan BM. Neurology. 2018.
20. Incidence, Etiology, and Long-Term Outcome of Acute Myelitis in Stockholm County, Sweden: A Population-Based Study. — Jonsson DI, Sveinsson O, Moeini N, et al. Neurology Neuroimmunology & Neuroinflammation. 2025.